New Insights Into the Evolution of Immune Adaptors in Murid Rodents
Qianqian Su, Zhenhua She, Yi Chen

TL;DR
This study explores how immune adaptors evolved in murid rodents, finding evidence of purifying selection and adaptive changes in specific genes.
Contribution
The study provides new insights into the molecular evolution of immune adaptors in murid rodents across multiple timescales.
Findings
Immune adaptors in murid rodents are mostly under purifying selection, with some positively selected sites in MAL and MyD88.
Genetic diversity in adaptor genes is reduced in Rattus tanezumi and Rattus norvegicus compared to noncoding regions.
Identical or nearly identical allelic variants were found between Rattus tanezumi and Rattus norvegicus in adaptor genes.
Abstract
The evolutionary dynamics of immune adaptors in wildlife remain poorly understood, despite their critical role in host–pathogen interactions. In this study, we investigated the molecular evolution of five TIR domain‐containing adaptors (MAL, MyD88, SARM, TRAM, and TRIF) in murid rodents across both interspecific and population levels to provide an integrative view of their evolutionary trajectories. Our analyses demonstrate that these adaptors are predominantly under purifying selection in murids, yet contain specific positively selected sites in MAL and MyD88, indicating localized adaptive fine‐tuning. These positively selected amino acid sites exhibited substantial genetic divergence among murid species. Within populations, we observed reduced genetic diversity in the adaptor genes compared with noncoding regions across the whole genome in both Rattus tanezumi and Rattus norvegicus…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
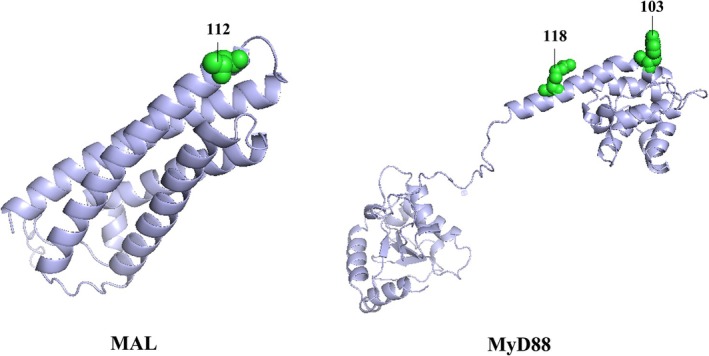 FIGURE 1
FIGURE 1 FIGURE 2
FIGURE 2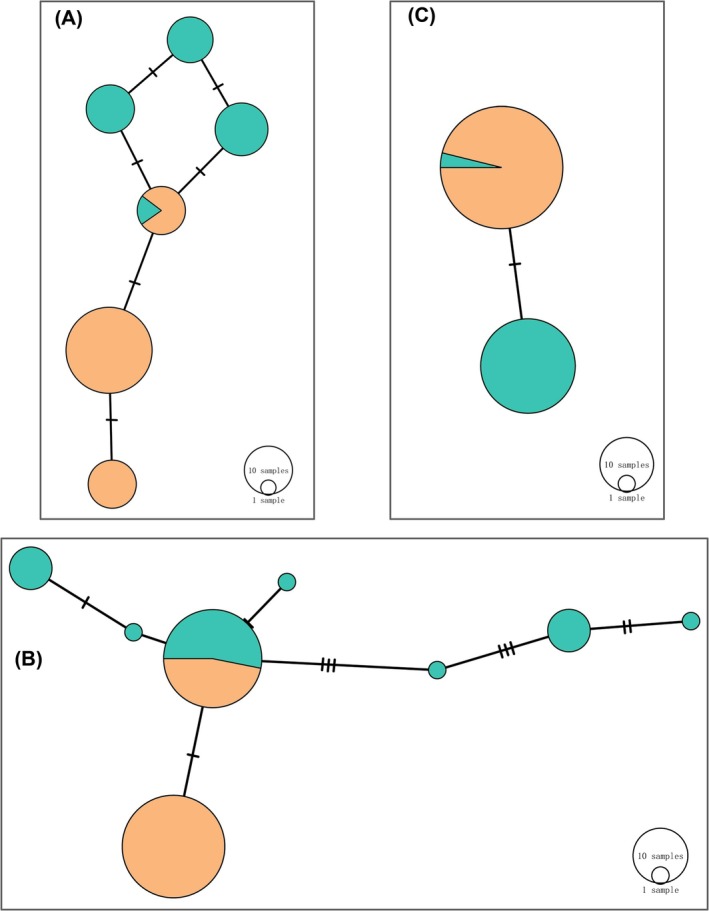 FIGURE 3
FIGURE 3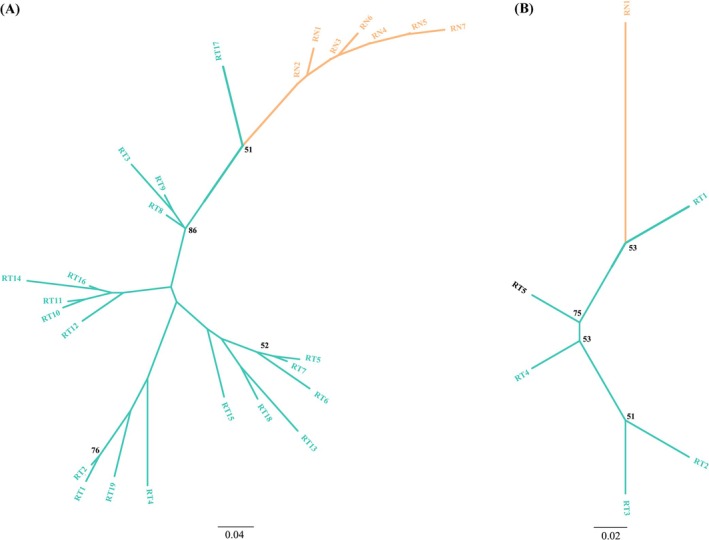 FIGURE 4
FIGURE 4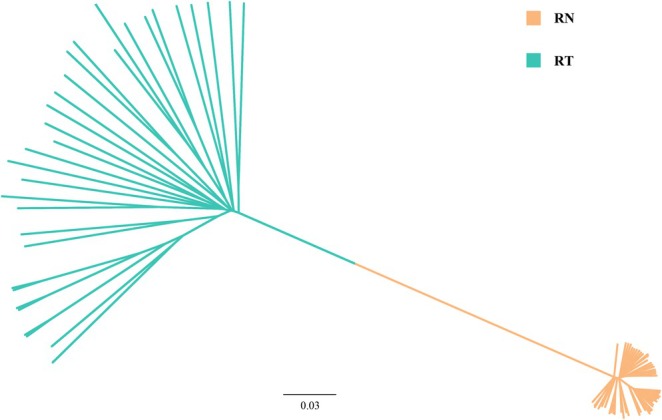 FIGURE 5
FIGURE 5| Gene | Function | Protein Accession number ( | References |
|---|---|---|---|
| MyD88 | Plays a central role in the innate and adaptive immune response and functions as an essential signal transducer in the interleukin‐1 and Toll‐like receptor signaling pathways |
| Horng et al. ( |
| MAL | Is essential for TLR2 and TLR4 signaling, functioning as a bridging molecule to recruit MyD88 |
| Horng et al. ( |
| TRIF | Plays a pivotal role in TLR3‐ and TLR4‐mediated pathways |
| Yamamoto et al. ( |
| TRAM1 | Contributes to the TLR4‐mediated MyD88‐independent pathway |
| Yammoto et al. ( |
| SARM1 | Negatively regulates TRIF‐dependent TLR signaling |
| Carty et al. ( |
| Species/Gene | Family | Genus | Myd88 | MAL | TRIF | TRAM1 | SARM1 |
|---|---|---|---|---|---|---|---|
|
| Muridae |
| |||||
|
| Muridae |
|
| # | # | # | # |
|
| Muridae |
| |||||
|
| Muridae |
| |||||
|
| Muridae |
| |||||
|
| Muridae |
| |||||
|
| Muridae |
| |||||
|
| Muridae |
| |||||
|
| Muridae |
| |||||
|
| Muridae |
| |||||
|
| Muridae |
| GCF_903995435.1* | GCF_903995435.1* | |||
|
| Muridae |
| GCF_907164565.1* | ||||
|
| Muridae |
| GCF_030254825.1* | GCF_030254825.1* | |||
|
| Muridae |
| GCA_003336285.2* |
| Gene | Number of species | Test of selection | Sites under selection identified by different methods | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| lnL M7 | lnL M8 | 2lnΔL | Significance |
| PAML M8 | FEL | SLAC | FUBAR | MEME | ||
| MAL | 12 | −1299.0858 | −1294.02013 | 10.1313 |
| 0.1752 |
|
|
| ||
| Myd88 | 13 | −3054.5642 | −3049.66584 | 9.7968 |
| 0.15148 | 39, | 49 |
| 12,68,94, | |
| TRAM1 | 14 | −3067.8741 | −3066.23595 | 3.276386 | ns | 0.04957 | 74 | 340 | |||
| TRIF | 12 | −1233.0372 | −1233.03861 | −0.002732 | ns | 0.14962 | |||||
| SARM1 | 13 | −5762.1405 | −2762.14184 | −0.0027 | ns | 0.02577 | 388 | ||||
| Gene | Size (bp) |
|
|
| Hd | π | θW | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RT | RN | RT | RN | RT | RN | RT | RN | RT | RN | RT | RN | ||
| MAL | 462 | 33 | 50 | 2 | 2 | 4 | 3 | 0.72 | 0.536 | 0.00212 | 0.00085 | 0.00107 | 0.00097 |
| MyD88 | 891 | 33 | 50 | 8 | 1 | 7 | 2 | 0.686 | 0.429 | 0.00329 | 0.00048 | 0.00221 | 0.00025 |
| SARM1 | 2175 | 33 | 50 | 17 | 4 | 19 | 7 | 0.955 | 0.811 | 0.00291 | 0.00084 | 0.00193 | 0.00041 |
| TRAM1 | 1125 | 33 | 50 | 3 | 0 | 5 | 1 | 0.708 | 0 | 0.00083 | 0 | 0.00066 | 0 |
| TRIF | 630 | 33 | 50 | 1 | 0 | 2 | 1 | 0.117 | 0 | 0.00019 | 0 | 0.00039 | 0 |
| Gene | Tajima's | Fu‐Li D* (average value of | Fu‐Li | MK test (NI value) | HKA test ( | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| RT | RN | RT | RN | RT | RN | RT | RN | RT | RN | |
| MAL | 0.0036 | −0.0491 | 0.0305 | −0.0734 | 0.0065 | −0.0721 | 1.2 | 0.001 | 1.731 | 0.001 |
| MyD88 | −0.1099 | −0.0218 | −0.0767 | −0.0338 | −0.0913 | 0.0174 | 0.883 | 0.266 | 0.749 | 1.23 |
| SARM1 | −0.1069 | −0.0723 | −0.0353 | −0.0383 | −0.0156 | −0.0436 | 0.5 | 2.9 | 0.523 | 0.892 |
| TRAM1 | −0.0892 | NA | −0.0273 | NA | −0.0176 | NA | 0.001 | NA | 1.79 | NA |
| TRIF | −0.0789 | NA | −0.0973 | NA | −0.0393 | NA | 0.001 | NA | 1.161 | NA |
- —Natural Science Foundation of Hunan Province10.13039/501100004735
- —Excellent Youth Project of Hunan Education Department
- —National Natural Science Foundation of China10.13039/501100001809
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsT-cell and B-cell Immunology · interferon and immune responses · Immune Response and Inflammation
Introduction
1
Animals face constant threats from pathogens and associated infectious diseases throughout their lifespans (Siddle and Quintana‐Murci 2014). The immune system functions as a highly complex defense system, in which receptors, adaptors, and effectors serve as core components, enabling it to detect, analyze, and eliminate threats. Receptors, such as Toll‐like receptors (TLRs), provide specificity by effectively identifying pathogenic infections. When TLRs bind to ligands from parasites or viruses, they typically do not directly trigger downstream responses; instead, adaptors act as signaling intermediates. Adaptors are responsible for signal transduction and the activation of immune responses. Effectors, as the ultimate executors of immune function, kill and eliminate threats, ultimately establishing immune memory against the pathogen (O'Neill et al. 2003). Population genetic studies have demonstrated that immune‐related genes are key targets of natural selection, underscoring the critical role of immune mechanisms in host defense and advancing our understanding of how organisms respond to selective pressures (Siddle and Quintana‐Murci 2014). Extensive evolutionary research has focused on receptors genes (Fitzgerald and Kagan 2020; Su et al. 2022, 2024). However, different components of the immune system may exhibit distinct evolutionary patterns. Evidence has shown that adaptors experience stronger selection pressure than TLRs in healthy individuals from diverse geographic regions, suggesting greater evolutionary constraints on maintaining adaptor protein integrity (Fornarino et al. 2011).
To date, five adaptor molecules have been identified in mammals (Table 1). The most well‐characterized adaptor is the myeloid differentiation primary response gene 88 (MyD88). The remaining four adaptors include MyD88‐adaptor‐like (MAL, also known as TIRAP), TIR domain‐containing adaptor inducing interferon‐β (TRIF; also designated TICAM1), TRIF‐related adaptor molecule (TRAM; TICAM2), and sterile α‐ and armadillo‐motif‐containing protein (SARM). MyD88 was the first adaptor discovered to mediate signaling across multiple TLRs. Critical evidence stems from MyD88‐deficient mice, which failed to respond to ligands for TLR2, TLR4, TLR5, TLR7, or TLR9 (Takeuchi et al. 2000; O'Neill and Bowie 2007; Vijay 2018). MAL, the second adaptor identified, is essential for TLR2 and TLR4 signaling, functioning as a bridging molecule to recruit MyD88 (Horng et al. 2001). TRIF plays a pivotal role in TLR3‐ and TLR4‐mediated pathways in mammals (Yamamoto et al. 2003). In TRIF‐deficient mice, both TLR3‐ and TLR4‐dependent IFN‐β expression and IRF3 activation were impaired (Yamamoto et al. 2003). TRAM specifically contributes to the TLR4‐mediated MyD88‐independent pathway, as TRAM‐deficient mice exhibit defective cytokine production in response to TLR4 ligands but not other TLR ligands (Yammoto et al. 2003). In contrast, SARM negatively regulates TRIF‐dependent TLR signaling (Carty et al. 2006). Overexpression of SARM suppresses downstream TRIF‐mediated gene induction but does not affect MyD88‐dependent signaling (Carty et al. 2006).
Previous work assessed the genetic diversity and evolutionary trajectories of these five TIR‐containing adaptors in human populations. MyD88 and TRIF exhibited signatures of purifying selection, consistent with their essential and nonredundant roles in early signal transduction (Fornarino et al. 2011). Additionally, multiple episodes of positive selection have shaped these adaptors, with distinct spatiotemporal patterns. Selective sweeps were detected in MyD88 and SARM across all human populations, whereas adaptive evolution in the other three adaptors was restricted to specific geographic groups (Fornarino et al. 2011). Nevertheless, the evolutionary history and genetic variation of these adaptors in wild mammals remain poorly understood, leaving significant gaps in our knowledge of their roles in natural populations.
In this study, we investigated genetic variation and molecular evolution of TIR‐containing adaptors in murid rodents (Rodentia: Muridae), the most widespread family of rats and mice. This family originated 20–30 million years ago, underwent rapid radiation into multiple subfamilies, and achieved a near‐global distribution (Musser and Carleton 2005). Their evolutionary success is evidenced by extensive adaptive radiation and global colonization (Puckett et al. 2016). Notably, murid rodents serve as asymptomatic reservoirs for at least 60 zoonotic pathogens (Baker et al. 2022). Commensal species like the Asian house rat ( Rattus tanezumi , RT) and brown rat ( Rattus norvegicus , RN) exhibit particularly close associations with human populations, facilitating transmission of pathogens including Hantavirus, Yersinia, Leptospira, Salmonella, and Bartonella (Wu et al. 2018; Su et al. 2022). This study addresses key knowledge gaps regarding TIR‐containing adaptor evolution in murid rodents. By characterizing evolutionary patterns in these species, we aim to enhance understanding of adaptor protein function in immune systems. Specifically, our objectives are to: (1) Determine evolutionary patterns of TIR‐containing adaptors across murid rodents through comparative genetic analysis; (2) Characterize evolutionary features of five adaptor genes in sympatric RT and RN populations using population genetic approaches.
Methods
2
Data Collection
2.1
We obtained full‐length sequences of five adaptor proteins from 14 different murid species from NCBI (https://www.ncbi.nlm.nih.gov/; accession numbers provided in Table 2). Attempts to extract DNA sequences from other murid species were partly unsuccessful due to low‐quality or incomplete genome data, which were consequently excluded. We performed sequence alignment using ClustalW (Codons) in MEGA6 (Tamura et al. 2013), with aligned sequences provided in Data S1–S5.
For population‐level analysis, we extracted all five adaptor sequences from whole‐genome data of RT (33 individuals) and RN (50 individuals) populations from our previous studies (NGDC accessions: CRA001635 and CRA003158, respectively; Chen, Hou, et al. 2021; Chen, Zhao, et al. 2021) (Table S1). First, we performed single‐nucleotide polymorphism (SNP) calling using the genome analysis toolkit (GATK v4.1) HaplotypeCaller protocol (McKenna et al. 2010). Then, we obtained adaptor gene sequences with GATK FastaAlternateReferenceMaker protocol (McKenna et al. 2010) with default parameters.
Interspecific Selection Analysis
2.2
Following established methodologies (Su et al. 2024), we employed multiple approaches to assess selection pressures: First, we used the maximum likelihood (ML) framework to evaluate positive and negative selection at each adaptor during Muridae evolution, based on the rate per site of nonsynonymous substitution (dN) to the rate per site of synonymous substitutions (dS). We used the neighbor joining trees as the working topology for the adaptor genes. When performing gene selection pressure analysis using PAML (particularly the codeml program), it is generally recommended to employ gene trees rather than species trees. If the topology of the gene tree conflicts with that of the species tree, imposing the species tree may lead to erroneous branch assignments, thereby affecting the estimation of the ω ratio (dN/dS) and potentially generating false‐positive or false‐negative outcomes. Because genes can undergo evolutionary histories distinct from the species as a whole, such as through positive selection acting on specific lineages, using a gene tree enables more accurate detection of such signals. For these reasons, gene trees were used for the PAML analysis in this study. We tested the effect of positive selection on Muridae lineage by estimating global ω values under the M7/M8 branch model with CODEML in PAML v4 (Yang 1997; Jeffares et al. 2015; Álvarez‐Carretero et al. 2023). The M7 model restricts codon evolution to either neutral evolution or purifying selection (where dN/dS ≤ 1), while the M8 model incorporates an additional category of sites evolving under positive selection (dN/dS > 1). These two nested models were compared using a likelihood ratio test (LRT) with 2 degrees of freedom (Nielsen and Yang 1998; Yang et al. 2000). Positively selected sites under model M8 were identified using a Bayes empirical Bayes (BEBs) approach (Yang et al. 2005). Sites with a posterior probability exceeding 90% were considered candidate targets of positive selection, as pinpointing individual selected sites is statistically more challenging than detecting the presence of selection among a proportion of sites (Wlasiuk and Nachman 2010; Těšický et al. 2020). The one‐ratio model (model M0, assuming the same ω for all branches and sites, model = 0 and Nsites = 0) analysis was also performed to test the overall selection pressure in Muridae species.
Next, we used several alternative selection tests to evaluate the selection pressure occurring in our diverse data set with slightly different assumptions and constraints. To do this, a series of ML methods were implemented in the DATAMONKEY Web server (Weaver et al. 2018). The fixed‐effect likelihood (FEL) model estimates the ratio of nonsynonymous to synonymous substitution on a site‐by‐site basis without assuming an a priori distribution of rates across sites (Kosakovsky Pond and Frost 2005). The single likelihood ancestor counting (SLAC) model is based on the reconstruction of ancestral sequences and the counts of synonymous and nonsynonymous changes at each codon position in a phylogeny (Kosakovsky Pond and Frost 2005). The fast, unconstrained Bayesian approximation (FUBAR) model is based on random effect likelihood methods (Murrell et al. 2013). The mixed effects model of evolution (MEME) employs a mixed‐effects ML approach to test the hypothesis that individual sites have been subjected to episodic positive or diversifying selection (Murrell et al. 2012). We performed all these analyses with the significance level of posterior probability established by default to 0.9. Codons were considered robust candidates for positive selection when they were identified to be under positive selection by at least two ML methods.
Population Genetic Analysis
2.3
We used DnaSP v5 to estimate the nucleotide polymorphism, haplotype number, nucleotide diversity, haplotype diversity (Hd), and average number of nucleotide differences in the gene sequences of these five adaptors with default parameters (Librado and Rozas 2009). We use “haplotype” to denote unique coding‐sequence alleles for each adaptor gene. We calculated the average nucleotide diversity values of noncoding regions across the whole genome using vcftools (Danecek et al. 2011). We built phylogenetic trees using MEGA 6 via the neighbor‐joining (NJ) method (Tamura et al. 2013). Additionally, we constructed phylogenetic trees based on all variant sites of the genome using VCF2Dis v1.42 (https://github.com/BGI‐shenzhen/VCF2Dis) with default arguments. We used Recombination Detection Program 4 (RDP4, Martin et al. 2015) and GENECONV v1.81 (Sawyer 1989) under default parameters to determine whether gene conversion occurred among rat populations for these adaptor genes.
Results
3
Genetic Characteristics at the Interspecific Level
3.1
Selection pressure analyses revealed that all five adaptor genes evolved under purifying selection (ω < 1; Table 3). Notably, we identified one (codon 112) and two (codon 103 and 118) positively selected amino acid sites in MAL and MyD88, respectively, which were consistently supported by multiple detection methods (Table 3; Figure 1). These three positively selected amino acid sites show high genetic diversity in murids (Figure 2).
Positively selected sites visualized on the three‐dimensional structures of MAL and MyD88. The structure and amino acids refer to Rattus norvegicus .
Details of mutations at positively selected amino acid sites (PSAs) among murid species. One PSA (codon 112) in MAL and two PSAs (codon 103 and 118) in MyD88.
Intraspecific Genetic Characteristics
3.2
Haplotype distributions differed significantly between RT and RN populations. The numbers of haplotypes of these five genes (MAL, MyD88, SARM1, TRAM1, and TRIF) in the RT population were 4, 7, 19, 5, and 2, respectively. For RN, the numbers of haplotypes of these five genes (MAL, MyD88, SARM1, TRAM1, and TRIF) were 3, 2, 7, 1, and 1, respectively. Both Hd and nucleotide diversity (π) were consistently higher in RT than RN for all genes (Table 4). Notably, the average nucleotide diversity values of the noncoding regions across the whole genome in the RT and RN populations were 0.00528 and 0.00088, respectively, both of which were greater than those of these five adaptor genes. The TRAM1 and TRIF genes showed complete conservation in RNs, and no mutation sites were detected. Moreover, we did not detect any significant signals of positive or negative selection in adaptor genes in either rat population (Table 5).
TABLE 4: Genetic characteristics for five adaptors in Rattus tanezumi and R. norvegicus populations.
TABLE 5: Netrality tests for five adaptors in Rattus tanezumi and R. norvegicus populations.
Surprisingly, identical or highly similar genetic variants (> 99.8% sequence identity) were observed between these two rat populations for all five of these adaptors. For the MAL, MyD88, and TRIF genes, one haplotype shared between RT and RN was observed (Figure 3). For the SARM1 and TRAM1 genes, at least one type of RT haplotype clustered together with the RN haplotypes (Figure 4). Conversely, phylogenetic trees based on genome‐scale data separated individuals of RTs and RNs completely (Figure 5). Moreover, we did not detect any recombination event between these two rat populations in adaptor genes.
Haplotype networks of MAL (A), MyD88 (B), and TRIF (C). The haplotype network was constructed in 83 rats including RT and RN. Circle size reflected the number of rats with the respective haplotype. A dash on the joined line indicated one nucleotide variation. Yellow color represent RN and green color represent RT.
Neighbor‐joining tree of SARM1 (A) and TRAM1 (B) genes from RT and RN populations. Branch lengths were computed based on p‐distance method. Each tip corresponds to a unique haplotype. Clades with yellow color represent RN haplotypes, and clades with green color represent RT haplotypes. Bootstrap percentages (from 1000 replications) for major clusters are shown on internal branches. Bootstraps values > 50 are indicated at their respective nodes. For SARM1, Haplotype RT17 clustered together with RN alleles (poor bootstrap support between them: 51%). For TRAM1, Haplotype RT1 clustered together with the RN allele (poor bootstrap support between them: 53%).
Neighbor‐joining tree based on all variant sites across the genome of RT and RN populations. Branch lengths were computed based on p‐distance method. Each tip corresponds to a unique haplotype. Clades with yellow color represent RN haplotypes, and clades with green color represent RT haplotypes.
Discussion
4
TIR‐domain‐containing adaptor proteins play pivotal roles in vertebrate immunity, making the study of their evolutionary patterns essential for ecological and evolutionary research. Our study provided novel insights into the evolutionary dynamics of TIR‐domain‐containing adaptor proteins in murid rodents, a highly successful mammalian family with global distribution. The predominant purifying selection observed across all five adaptors (ω < 1) underscored their essential and conserved roles in TLR‐mediated immune responses (O'Neill and Bowie 2007). This evolutionary pattern aligned with previous findings in humans (Fornarino et al. 2011), suggesting strong functional constraints on these critical signaling molecules throughout mammalian evolution.
The identification of positively selected sites in MyD88 (positions 103 and 118) and MAL (position 112) Present particularly intriguing findings. The MyD88 variants occur in functionally critical domains: position 103 within the death domain (DD) that mediates myddosome assembly through IRAK interactions (Ve et al. 2017), and position 118 in the intermediary domain essential for proper protein conformation (Avbelj et al. 2011). Their location in key functional domains suggests potential adaptive significance, which needs to be validated experimentally in future studies. Previous studies have demonstrated that analogous mutations in these domains can significantly alter signaling outcomes (Loiarro et al. 2009; Ve et al. 2017). The positively selected site in MAL's TIR domain (position 112) may represent another adaptive hotspot, particularly given established evidence that TIR domain variants can dramatically impact TLR2/4 signaling (Nagpal et al. 2009). The known association between MAL polymorphisms and infectious disease resistance in humans (Khor et al. 2007) further supports the potential functional importance of this variation. These findings collectively suggest that while adaptor proteins are generally under strong purifying selection, certain residues may undergo positive selection to fine‐tune immune responses in specific ecological contexts. The remarkable conservation of these adaptors in murid rodents, particularly when compared to their neutral region diversity, highlights their fundamental importance in maintaining immune system integrity. This conservation is especially noteworthy given the exceptional adaptability of murid rodents to diverse environments and their role as reservoirs for numerous zoonotic pathogens (Wu et al. 2018; Su et al. 2022). The observed evolutionary patterns may reflect balancing selection pressures to maintain robust pathogen recognition while avoiding excessive immune activation. Specially, our findings raise several important questions for future research. First, experimental studies are needed to characterize the functional consequences of the identified variants. Second, comparative analyses across additional mammalian taxa could reveal whether these patterns represent rodent‐specific adaptations or more general evolutionary trends.
Our study revealed striking genetic similarity in five adaptor proteins between sympatric RT and RN populations. Such shared polymorphisms in closely related species typically arise through four evolutionary mechanisms: (1) recombination, (2) trans‐species polymorphism (TSP), (3) convergent evolution, or (4) incomplete lineage sorting (ILS) (Cho et al. 2006; Těšický and Vinkler 2015). Recombination was not detected between these two rat populations in adaptor genes. TSP is generally observed in highly polymorphic immune genes like MHC and TRIM5α, where balancing selection maintains ancestral variants across species (Cagliani et al. 2010; Azevedo et al. 2015; Million and Lively 2022). The adaptor genes showed low polymorphism in both RT and RN populations. However, TSPs may not require high diversity when they involve a small number of long‐lived alleles. Future study should be conducted with broader outgroup sampling to reconstruct the ancestral state of these rat species. Convergent evolution refers to the independent evolution of similar phenotypic features or functions among different taxa with distant genetic relationships, due to adaptation to similar environments or ecological niches, under the influence of natural selection. These features do not arise from a common direct ancestor, but from independent origins (McGhee 2011; Stayton 2015). ILS refers to the phenomenon of “gene tree and species tree inconsistency” that occurs at the genetic level in rapidly differentiated species in recent times. The reason is that during species differentiation, certain genes in the ancestral population exhibit polymorphism (multiple alleles), and subsequently different alleles are randomly fixed in different offspring lineages, resulting in a mismatch between the evolutionary relationship of certain genes and the true differentiation relationship of the species (Degnan and Rosenberg 2009). RT and RN arose from the same lineage (Rattus), and the divergence time between these two species is less than 3 million years (Robins et al. 2008). Moreover, no significant signals of recent positive or negative selection were detected in either rat population, indicating the weak natural selection acting on adaptor genes of these two rat populations. Therefore, compared to convergent evolution, TSP/ILS is more likely to be the cause of the similarity and shared haplotypes in adaptor genes between these two rat populations.
Conclusions
5
This study significantly advances our understanding of immune gene evolution in ecologically important species and provides a foundation for future investigations into the molecular mechanisms underlying pathogen–host interactions in natural populations. The combination of evolutionary conservation and targeted diversification at key residues suggests a sophisticated balance between maintaining core immune functions and allowing adaptive fine‐tuning in these critical signaling molecules.
Author Contributions
Qianqian Su: data curation (equal), formal analysis (equal), funding acquisition (equal), investigation (equal), methodology (equal), project administration (equal), visualization (equal), writing – original draft (equal). Zhenhua She: methodology (equal). Yi Chen: conceptualization (equal), formal analysis (equal), methodology (equal), resources (equal), validation (equal), writing – review and editing (equal).
Funding
This work was supported by Natural Science Foundation of Hunan, 2023JJ41038. Excellent Youth Project of Hunan Education Department, 22B0257. National Natural Science Foundation of China, 32400413.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Data S1: ece372851‐sup‐0001‐DataS1.docx.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Álvarez‐Carretero, S. , P. Kapli , and Z. Yang . 2023. “Beginner's Guide on the Use of PAML to Detect Positive Selection.” Molecular Biology and Evolution 40, no. 4: msad 041.37096789 10.1093/molbev/msad 041PMC 10127084 · doi ↗ · pubmed ↗
- 2Avbelj, M. , S. Horvat , and R. Jerala . 2011. “The Role of Intermediary Domain of My D 88 in Cell Activation and Therapeutic Inhibition of TL Rs.” Journal of Immunology 187, no. 5: 2394–2404.10.4049/jimmunol.110051521804016 · doi ↗ · pubmed ↗
- 3Azevedo, L. , C. Serrano , A. Amorim , et al. 2015. “Trans‐Species Polymorphism in Humans and the Great Apes Is Generally Maintained by Balancing Selection That Modulates the Host Immune Response.” Human Genomics 9: 1–6.26337052 10.1186/s 40246-015-0043-1PMC 4559023 · doi ↗ · pubmed ↗
- 4Baker, R. E. , A. S. Mahmud , I. F. Miller , et al. 2022. “Infectious Disease in an Era of Global Change.” Nature Reviews Microbiology 20, no. 4: 193–205.34646006 10.1038/s 41579-021-00639-z PMC 8513385 · doi ↗ · pubmed ↗
- 5Cagliani, R. , M. Fumagalli , M. Biasin , et al. 2010. “Long‐Term Balancing Selection Maintains Trans‐Specific Polymorphisms in the Human TRIM 5 Gene.” Human Genetics 128: 577–588.20811909 10.1007/s 00439-010-0884-6 · doi ↗ · pubmed ↗
- 6Carty, M. , R. Goodbody , M. Schröder , J. Stack , P. N. Moynagh , and A. G. Bowie . 2006. “The Human Adaptor SARM Negatively Regulates Adaptor Protein TRIF‐Dependent Toll‐Like Receptor Signaling.” Nature Immunology 7, no. 10: 1074–1081.16964262 10.1038/ni 1382 · doi ↗ · pubmed ↗
- 7Chen, Y. , G. Hou , M. Jing , et al. 2021. “Genomic Analysis Unveils Mechanisms of Northward Invasion and Signatures of Plateau Adaptation in the Asian House Rat.” Molecular Ecology 30, no. 24: 6596–6610.34564921 10.1111/mec.16194 · doi ↗ · pubmed ↗
- 8Chen, Y. , L. Zhao , H. Teng , et al. 2021. “Population Genomics Reveal Rapid Genetic Differentiation in a Recently Invasive Population of Rattus norvegicus .” Frontiers in Zoology 18: 1–10.33499890 10.1186/s 12983-021-00387-z PMC 7836188 · doi ↗ · pubmed ↗