First LDLRAP1 and Recurrent LDLR Mutations in Tunisian Families With Familial Hypercholesterolemia
Wirath Ben Ncir, Afif Ben‐Mahmoud, Hamdi Frikha, Fatma Abdelhedi, Faten Hadj Kacem, Nabila Majdoub, Mouna Mnif, Hyung‐Goo Kim, Leila Ammar Keskes, Jouke‐Jan Hottenga

TL;DR
This study reports the first cases of autosomal recessive familial hypercholesterolemia in Tunisia, caused by mutations in the LDLRAP1 gene, and highlights the genetic diversity of the condition.
Contribution
The first identification of LDLRAP1 mutations in Tunisian families with autosomal recessive familial hypercholesterolemia.
Findings
A novel homozygous LDLRAP1 missense variant (p.Gly54Asp) was found to cause autosomal recessive FH in one family.
A recurrent LDLR splice-site variant was identified in another family, following an autosomal dominant inheritance pattern.
The LDLRAP1 variant was predicted to destabilize the PTB domain, likely impairing LDL receptor internalization.
Abstract
Familial hypercholesterolemia (FH) is a genetic disorder characterised by elevated plasma LDL‐cholesterol, predisposing to premature atherosclerotic cardiovascular disease. Most cases follow an autosomal dominant pattern (ADH) caused by pathogenic variants in LDLR, APOB or PCSK9. In contrast, the rare autosomal recessive form (ARH) results from biallelic mutations in LDLRAP1, leading to defective LDL receptor‐mediated endocytosis. Despite the high rate of consanguinity in Tunisia, LDLRAP1 variants have not yet been reported in this population. In this study, Whole Exome Sequencing of two consanguineous Tunisian families, identified distinct pathogenic variants. In the first family (FH‐A), a recurrent LDLR splice‐site variant (c.1845+1G>A) was detected in both heterozygous and homozygous states, consistent with an autosomal dominant inheritance pattern. In the second family (FH‐B), a…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
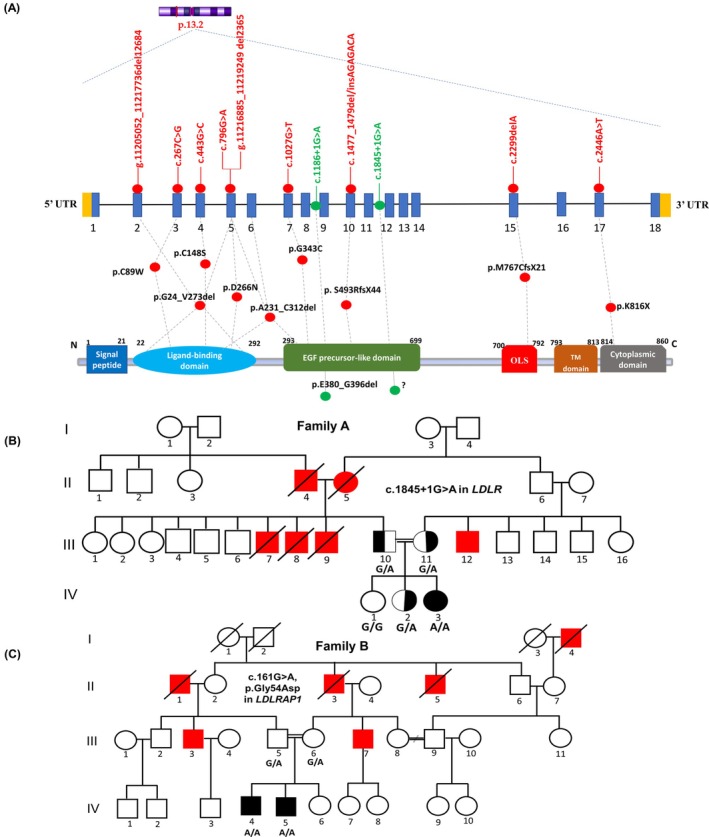 FIGURE 1
FIGURE 1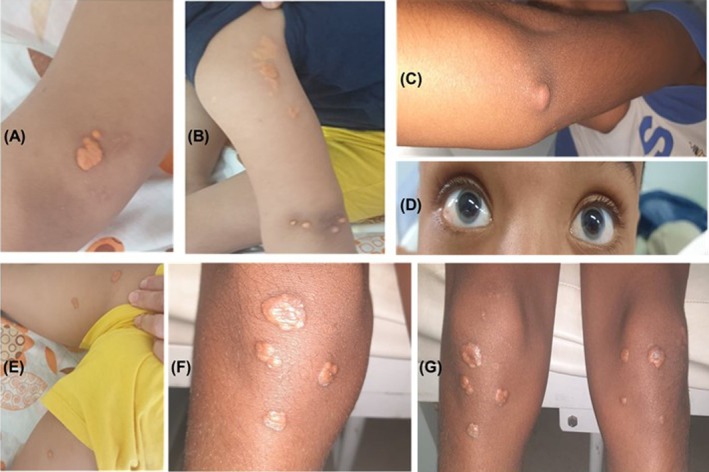 FIGURE 2
FIGURE 2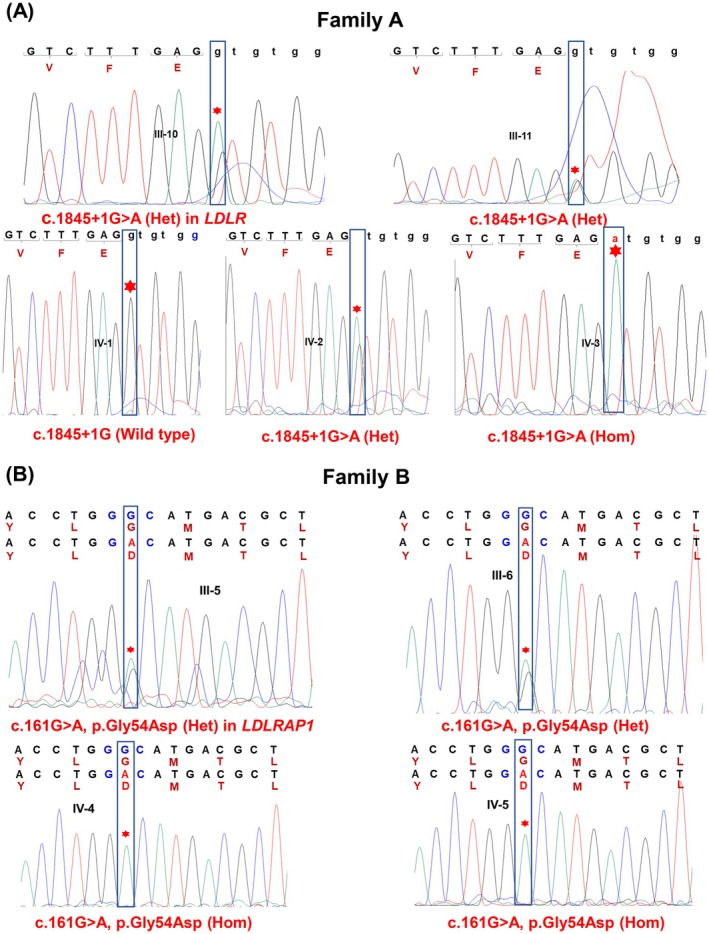 FIGURE 3
FIGURE 3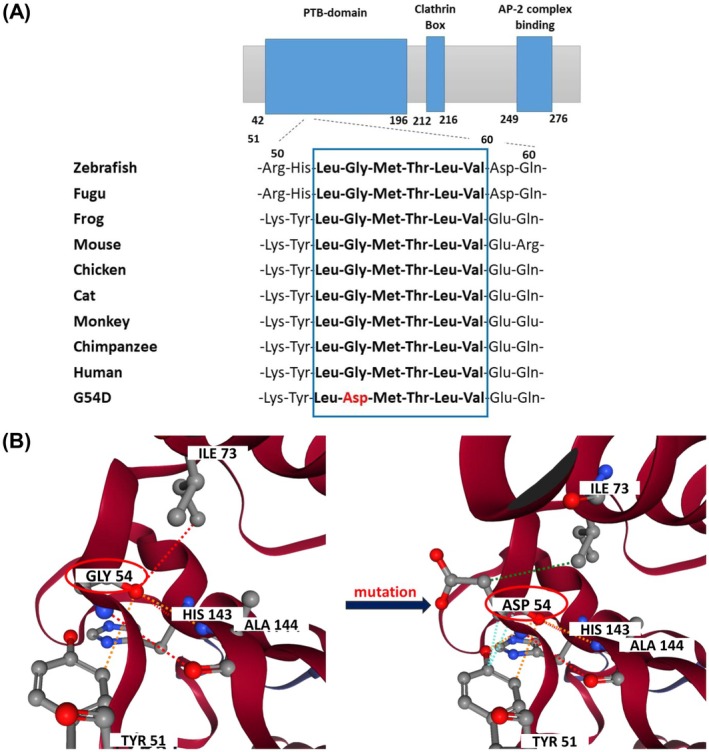 FIGURE 4
FIGURE 4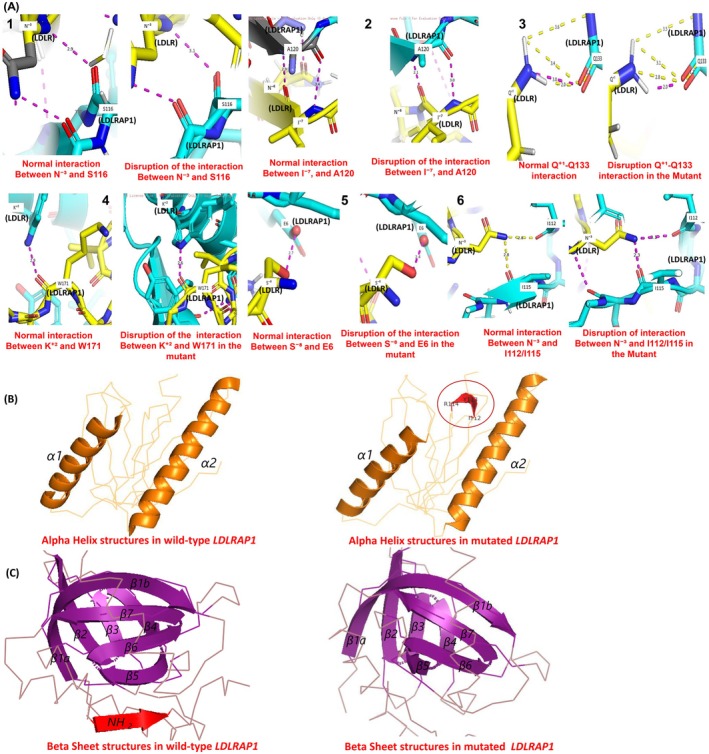 FIGURE 5
FIGURE 5| Exon/intron | cDNA variant | Protein variant | Variant type | References |
|---|---|---|---|---|
|
| ||||
| 2_5 | g.11205052_11217736del12685 | p.Gly24_Val 273del | Major rearrangement | [ |
| 3 | c.267C>G | p.Cys89Trp | Missense | [ |
| 4 | c.443G>C | p.Cys148Ser | Missense | [ |
| 5 | c.796G>A | p.Asp266Asn | Missense | [ |
| 5_6 | g.11216885_11219249del2365 | p.Ala231_Cys312del | Major rearrangement | [ |
| 7 | c.1027G>T | p.Gly343Cys | Missense | [ |
| 8–9 | c.1186+1G>A | p.Glu380_Gly396del | Splice site | [ |
| 10 | c.1477_1479del/insAGAGACA | p.Ser493ArgfsX44 | Frame shift | [ |
| 11/12 | c.1845+1G>A | ? | Splice site | [ |
| 15 | c.2299delA | p.Met767CysfsX21 | Frameshift | [ |
| 17 | c.2446A>T | p.Lys816X | Nonsense | [ |
|
| ||||
| 3 | c.520C>T | p.Pro174Ser | Missense | [ |
| Family ID | Code | Sex | Age | TC mmol/L | TG mmol/L | LDL‐C mmol/L | HDL‐C mmol/L | Tuberous xanthomas | Tendinous xanthomas | Corneal acrus | CHD | Treatment |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | III‐10 | M | − | 5.9 | 1.7 | 4.3 | 0.8 | + | Atorvastatin 80 mg/day+ ezetemibe 10 mg/day | |||
| III‐11 | F | − | 9.3 | 1.43 | 7.3 | 1.33 | − | Atorvastatin 20 mg/day | ||||
| IV‐1 | F | − | Rosuvastatin 5 mg/day | |||||||||
| IV‐2 | F | 8 years | 8.7 | 0.67 | 7.08 | 1.31 | − | − | ||||
| IV‐3 | F | 6 years | 28.4 | 1.6 | 26.37 | 1.3 | + | + | + | − | Atorvastatin 10 mg/day | |
| B | III‐5 | M | — | Normolipidemic | ||||||||
| III‐6 | F | — | Normolipidemic | |||||||||
| IV‐4 | M | 12 years | 7.4 | 0.78 | 5.75 | 1.3 | + | + | + | − | Rosuvastatin 10 mg/day | |
| IV‐5 | M | 9 years | 8.47 | 0.8 | 6.90 | 1.21 | + | + | − | Rosuvastatin 10 mg/day | ||
- —Qatar Biomedical Research Institute, Hamad Bin Khalifa University10.13039/100019706
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLipoproteins and Cardiovascular Health · Blood disorders and treatments · Diabetes, Cardiovascular Risks, and Lipoproteins
Introduction
1
Familial hypercholesterolemia (FH) is an inherited disorder characterised by elevated serum low‐density lipoprotein cholesterol (LDL‐C), leading to premature atherosclerosis and cardiovascular disease. Excess LDL‐C leads to cholesterol deposition in peripheral tissues (xanthomas) and accelerates atherosclerosis, promoting early‐onset coronary heart disease [1]. FH is primarily caused by pathogenic variants in genes regulating LDL metabolism—LDLR, APOB, PCSK9, and, more rarely, LDLRAP1 [2]. Among these, LDLR mutations account for nearly 90% of cases, typically inherited in an autosomal dominant manner [3].
At the molecular level, the LDLR gene encodes the low‐density lipoprotein receptor, a transmembrane glycoprotein responsible for binding and internalising circulating LDL particles via apolipoprotein B. Once internalised through clathrin‐coated pits, the receptor‐ligand complex undergoes endocytosis, followed by dissociation in the endosome and subsequent recycling of the receptor to the cell surface. LDLRAP1 encodes a cytosolic adaptor protein essential for linking LDLR to the clathrin and AP‐2 complex, ensuring efficient LDL uptake [4, 5].
Autosomal recessive hypercholesterolemia (ARH) is caused by pathogenic variants in LDLRAP1, which disrupt LDL internalisation without altering the receptor structure, despite normal LDLR expression [6, 7]. In contrast, autosomal dominant hypercholesterolemia (ADH) results from mutations in LDLR, APOB or PCSK9, which directly impair LDL binding, ligand integrity or receptor availability at the cell surface. This defect typically results in plasma LDL‐C levels exceeding 500 mg/dL and confers a high risk of early‐onset complications, including atherosclerotic cardiovascular disease and aortic valve stenosis [4, 8]. Studies using patient‐derived induced pluripotent stem cell (iPSC)‐derived hepatocyte‐like cells have shown that LDLRAP1 deficiency only partially reduces LDL uptake, suggesting the existence of alternative internalisation pathways [9]. In addition to the classical FH‐associated genes, ApoE variants have also been implicated. ApoE, a key component of VLDL and chylomicrons, mediates hepatic lipoprotein uptake via receptors including LDLR. Pathogenic ApoE variants can disrupt this process, leading to a phenotype similar to autosomal dominant FH, overall contribution is relatively minor compared with the canonical FH genes [10, 11, 12].
Heterozygous familial hypercholesterolemia (HeFH) affects about 1 in 311 individuals, whereas the homozygous form (HoFH), caused by biallelic pathogenic variants in LDLR, APOB, PCSK9 or LDLRAP1, is far rarer, with an estimated prevalence of 1 in 250,000–360,000 [13]. The frequency varies by ethnicity, being highest in Black populations and lowest in Asian populations [3, 10, 14, 15, 16, 17]. Early diagnosis is crucial, as up to 90% of affected individuals remain undiagnosed and fewer than 1% receive adequate treatment [18]. Genetic testing allows accurate distinction between HeFH and HoFH, enabling timely initiation of intensive lipid‐lowering therapy, particularly in children at risk of premature cardiovascular disease [19].
In Tunisia, the prevalence of HeFH is estimated at approximately 1 in 165, significantly higher than the global average [20]. Nevertheless, systematic screening and molecular characterisation remain limited. To date, eleven LDLR and one PCSK9 pathogenic variants have been reported, while no LDLRAP1 variants have been identified (Figure 1A and Table 1) [21, 22, 23, 24, 25, 26, 27, 28]. This highlights a major gap in the understanding of genetic spectrum of FH within the Tunisian population.
Genetic and clinical characterisation of Tunisian families with familial hypercholesterolemia. (A) Schematic representation of Reported LDLR Mutations in Tunisian Patients at both the cDNA and Protein Levels. The upper panel shows LDLR gene located at 19p13.2, illustrating the 11 intragenic mutations (red circles) and two splice‐site variants (green circles) mapped to their corresponding exons and introns (LDLR, NM_000527.5). Exons are depicted in blue boxes, introns as connecting black lines, and untranslated regions (UTRs) as yellow boxes. The lower panel illustrates the LDLR protein domain structure (NP_000518.1), including the signal peptide (exon 1), ligand‐binding domain (exons 2–6), EGF precursor‐like domain (exons 7–14), O‐linked sugar (OLS) domain (exon 15), transmembrane (TM) domain (exons 16 and 5′ part of exon 17), and the cytoplasmic domain (3′ region of exon 17 and 5′ region of exon 18). Red and green dots beneath the protein structure indicate the precise locations of identified variants. (B, C) Pedigrees of consanguineous Tunisian families A and B carrying LDLR and LDLRAP1 variants, respectively. Red symbols indicate individuals diagnosed with cardiovascular disease (CVD) before the age of 50. A diagonal line denotes deceased individuals, and shaded symbols represent affected genotyped members.
The present study aims to investigate the molecular basis of FH in two consanguineous Tunisian families, identify causative variants and inheritance patterns, and expand knowledge of FH genetics in North Africa. Tunisia's unique genetic diversity—shaped by centuries of admixture among Arab, Berber, European and Sub‐Saharan ancestries—may contribute to a distinctive distribution of FH‐associated variants, underscoring the need for population‐specific studies.
Material and Methods
2
Patients
2.1
Nine individuals from two unrelated Tunisian families (A and B) were analysed, including three probands and six first‐degree relatives. The pedigrees of families A and B are shown in Figure 1B,C, respectively.
The probands were diagnosed with FH based on established clinical criteria, including markedly elevated plasma levels of total cholesterol and low‐density lipoprotein cholesterol (LDL‐C > 8 mmol/L), a family history of hypercholesterolemia, and the presence of characteristic dermatological signs such as tuberous xanthomas (particularly on the elbows and knees), tendon xanthomas and corneal arcus (gerontoxon). These cutaneous signs were first noted during dermatological examinations (Figure 2).
Clinical presentation of cutaneous and ocular manifestations in patients from Families A and B. Representative clinical features of affected individuals IV‐3 (Family A; panels A, B and E) and IV‐4 (Family B; panels C, F and G). Multiple well‐demarcated yellowish plaques and nodules consistent with xanthomas are visible on the elbows (A–C), axilla (B), groin (E) and knees (F, G). Subcutaneous nodules are present on the extensor surface of the arm (B). An ocular abnormality, corneal arcus, is evident in both eyes of patient IV‐4 (D).
Further evaluation revealed diffuse carotid artery infiltration with atheromatous plaques in the right common carotid artery. Personal and/or familial histories of premature cardiovascular disease were also documented. Secondary causes of hypercholesterolemia, including diabetes mellitus, hypothyroidism and nephrotic syndrome, were systematically excluded.
Detailed family and medical histories were obtained for each subject following informed consent. The families were selected based on the Dutch Lipid Clinic Network (DLCN) criteria, a widely used tool for the clinical diagnosis of FH. This method integrates clinical and family histories, physical examination and LDL‐C levels. A DLCN score of > 6 was considered indicative of probable or definite FH, in accordance with current international guidelines [3, 13]. The lipid profile, additional clinical diagnoses and treatment regimens of the patients are summarised in Table 2.
Genetic Analysis and Predicting of Variant Pathogenicity
2.2
Genomic DNA was extracted from frozen whole‐blood samples collected in EDTA tubes using the standard phenol–chloroform method. DNA quantification was then performed using a Qubit fluorometer, and its purity was assessed using a NanoDrop spectrophotometer. Following quality validation, Whole Exome Sequencing (WES) was performed on DNA samples from all individuals included in this study, as previously described [29].
The bioinformatics pipeline for WES data analysis involved sequential steps of variant filtration and prioritisation. Analysis was restricted to exonic regions and canonical splice sites regions. Variants with a minor allele frequency (MAF) < 1% in population databases (gnomAD) were retained. Variants were then prioritised according to the known involvement of the corresponding gene in familial hypercholesterolemia and the observed co‐segregation of the variant with the disease within affected families. All candidate variants were cross‐referenced with dbSNP and ClinVar to identify previously reported variants and to determine any known clinical significance.
Variants interpretation followed ACMG guidelines and classified as pathogenic, likely pathogenic, variant of uncertain significance (VUS), likely benign, or benign [30]. Candidate variants implicated in FH causation were subsequently confirmed by Sanger sequencing. Specific primers were designed to amplify exon 2 of LDLRAP1 and exon 12 of LDLR via polymerase chain reaction (PCR) [30]. Primer sequences and annealing temperatures are provided in Table S1. PCR products were sequenced using an Applied Biosystems Genetic Analyser, and forward DNA strands of the targeted exons were analysed.
To evaluate the functional consequences of candidate variants, several in silico prediction tools were used, including MutationTaster, Meta‐SNP, CADD and MutPred2. MutationTaster assesses both coding and non‐coding variants by integrating evolutionary conservation, splice‐site alterations, protein features and known disease mutations. Meta‐SNP is a meta‐predictor that combines outputs from PANTHER, PhD‐SNP, SNAP and SIFT to generate a consensus pathogenicity score. MutPred2 predicts the structural and functional effects of non‐synonymous substitutions using sequence‐based, structural and evolutionary parameters. CADD (Combined Annotation Dependent Depletion) integrates multiple genomic annotations to provide a Phred‐like score ranking variants by their likelihood of pathogenicity.
Comparative Analysis of Amino Acid Conservation
2.3
The reference protein sequence of LDLRAP1 was retrieved from UniProt (accession number Q5SW96). For structural reference, the Protein Data Bank (PDB) entry corresponding to the LDLR cytoplasmic tail bound to the phosphotyrosine‐binding (PTB) domain of LDLRAP1 was obtained from the RCSB PDB database (accession number 3SO6).
Comparative sequence analysis of LDLRAP1 orthologs across multiple species was performed to evaluate amino acid conservation. Reference sequences were retrieved from Ensembl (https://www.ensembl.org) for Danio rerio (zebrafish), Takifugu rubripes (fugu), Xenopus tropicalis (frog), Mus musculus (mouse), Gallus gallus (chicken), Felis catus (cat), Macaca mulatta (monkey), Pan troglodytes (chimpanzee) and Homo sapiens (human) (Figure 4A).
Evaluation of Protein Stability and Dynamics
2.4
To assess the effects of amino acid substitutions on protein stability and dynamics, three computational tools were employed: DynaMut2, MUpro and DDGun [31, 32]. DynaMut2 applies normal mode analysis (NMA) to estimate variant‐induced changes in protein flexibility and stability, providing graphical visualisation of conformational alterations [33]. MUpro integrates both sequence‐based and structure‐based predictive models to evaluate the impact of variants on protein folding, stability and function. DDGun combines sequence‐derived features and evolutionary conservation data to predict the effects of single amino acid substitutions on protein stability. Collectively, these tools enhance the interpretation of structural consequences of variants and support the identification of potentially pathogenic variants.
Structural Analysis and Functional Prediction
2.5
To further evaluate the structural impact of the identified pathogenic variant, several computational tools were employed. NetSurfP‐2.0 was used to predict secondary structure, residue disorder and solvent accessibility at the mutation site. This tool employs an advanced architecture combining convolutional neural networks (CNNs) and long short‐term memory (LSTM) networks for highly accurate structural predictions [34]. AlphaFold2, was applied to predict the three‐dimensional (3D) conformation of proteins with high accuracy. The AlphaMissense extension of AlphaFold2 was used to predict the potential pathogenicity of missense variants [35]. Additionally, ConSurf was used to assess evolutionary conservation of residues at and around the mutation site [36].
Protein–Protein Interactions and Structural Properties Analysis
2.6
To investigate the interactions between LDLRAP1 and LDLR, the protein complex structure (PDB ID: 3SO6) was retrieved from the Protein Data Bank. Using the PyMOL mutagenesis wizard, the specific variant was introduced at the relevant residue within the phosphotyrosine‐binding (PTB) domain of LDLRAP1. PyMOL tools were then employed to compare the binding interfaces and interactions between LDLRAP1 and LDLR in both the wild‐type and mutant structures. Structural alterations, including changes in β‐sheet and α‐helix elements, were analysed and visualised using PyMOL graphics software to generate detailed figures illustrating these differences [37].
Results
3
Identification of the c.1845+1G>A Mutation in
LDLR in Tunisian Family A
3.1
WES identified a rare co‐segregating variant in the LDLR gene in the first Tunisian family with FH (Family A): c.1845+1G>A, corresponding to a guanine‐to‐adenine substitution at the +1 position of intron 12. This splice‐site variant alters the highly conserved GT dinucleotides at the 5′ donor site to AT, a change predicted to disrupt normal mRNA splicing. Sanger sequencing revealed that the affected proband (IV‐3) from family A is homozygous for the LDLR splice‐site variant c.1845+1G>A mutation in intron 12 (NM_000527.5; Figures 1B and 3A). The mother (III‐11), father (III‐10) and sister (IV‐2), all affected, are heterozygous carriers, whereas the unaffected sister (IV‐1) carried no variant allele (Figures 1B and 3A). Segregation analysis is consistent with an autosomal dominant inheritance in this family.
Sanger sequencing chromatograms showing wild‐type and mutant alleles of LDLR and LDLRAP1 in Tunisian families with familial hypercholesterolemia. Red asterisks indicate the positions of the nucleotide substitutions. (A) Splice‐site mutation c.1845+1G>A in LDLR identified in Family A. Individuals III‐10 and III‐11 (parents) are heterozygous carriers; IV‐1 (unaffected daughter) is homozygous wild‐type; IV‐2 is heterozygous; and IV‐3 (affected proband) is homozygous for the variant. Lowercase letters represent intronic sequences, uppercase letters denote exonic sequences, and the corresponding amino (in brown) are shown below the exonic codons. (B) Missense variant c.161G>A (p.Gly54Asp) in LDLRAP1 identified in Family B. Above each chromatogram, the wild‐type (top) and mutant (bottom) nucleotide and amino acid sequences are displayed. Individuals III‐5 and III‐6 (unaffected parents) are heterozygous carriers, while IV‐4 and IV‐5 (affected brothers) are homozygous for the variant. Variant nomenclature follows HGVS recommendations.
Identification of the c.161G>A Variant in
LDLRAP1 in Tunisian Family B
3.2
WES performed on four members from the second Tunisian family with (family B) identified a novel co‐segregating missense variant, c.161G>A, in exon 2 of the LDLRAP1 gene (NM_015627.3) (Figures 1C and 3B). This variant was present in the homozygous state in the two affected brothers (IV‐4 and IV‐5) and in the heterozygous state in their unaffected consanguineous parents (III‐5 and III‐6) (Figures 1C and 3B).
The LDLRAP1 c.161G>A variant results in Gly54Asp amino acid substitution (NP_056442.2). This residue is located within the N‐terminal PTB domain, which mediates the interactions of LDLRAP1 with the FDNPxY motif in the cytoplasmic tail of the LDL receptor and with phosphoinositides at the cell membrane. Functionally, LDLRAP1 also associates with clathrin and its adapter AP‐2 via its C‐terminus sequences, thereby facilitating LDLR internalisation through clathrin‐mediated endocytosis.
Based on the ACMG criteria, this variant was classified as likely pathogenic. According to multiple in silico prediction tools (see Methods), the p.Gly54Asp substitution is predicted to impair protein stability and function. Sanger sequencing confirmed the segregation pattern observed in the WES analysis (Figures 1C and 3B).
Evolutionary Conservation of the Gly54 Residue in
LDLRAP1 Across Vertebrate Orthologs
3.3
Multiple sequence alignment of LDLRAP1 with nine orthologs revealed that Gly54 is fully conserved across a wide range of mammalian and non‐mammalian species (Figure 4A).
Evolutionary conservation and structural impact of the LDLRAP1 p.Gly54Asp variant. (A) Multiple sequence alignment of LDLRAP1 across nine vertebrate species showing strong evolutionary conservation of Gly54, along with five additional residues highlighted in the blue box. (B) Three‐Dimensional Structural Visualisation of LDLRAP1 Wild‐Type and p.Gly54Asp Variant proteins generated using DynaMut2. Dotted lines indicate molecular interactions: Red for hydrogen bond, orange for polar bond, green for hydrophobic interactions, sky blue for VDW contacts. In the wild‐type protein, Gly54 forms hydrogen bonds with Ile73 and Ala144, stabilising the local structure. In the p.Gly54Asp variant, these hydrogen bonds are lost. The substitution Asp54 instead forms a hydrogen bond with His143, a hydrophobic interaction with Ile73, and a Van der Waals contact with Tyr51. These changes in intra‐protein interactions introduce steric hindrance, potentially disrupting proper protein folding and compromising LDLRAP1 function.
In Silico Structural and Functional Characterisation of the LDLRAP1 p.Gly54Asp Variant
3.4
The potential structural and functional impact of the novel LDLRAP1 c.161G>A (p.Gly54Asp) variant was assessed using multiple in silico prediction tools, including MutationTaster, Meta‐SNP, MutPred2 and CADD. Given the crucial role of LDLRAP1 in mediating LDL uptake by hepatocytes, computational evaluation of this variant is essential. Predictive analyses consistently indicated a deleterious effect: MutationTaster classified the variant as likely pathogenic, supported by a PhastCons score of 1 and PhyloP values of 5.908 and 1.503, reflecting strong evolutionary conservation. MutPred2 assigned a high pathogenicity score (0.940; p = 0.04) and predicted a gain of acetylation at lys51 and potential disruptions in functional motifs listed in the ELM (Eukaryotic Linear Motif) database (e.g., ELME000064, ELME000106, ELME000120), suggesting interference with critical interaction sites. Additionally, the variant obtained a CADD score of 32 (chr1:25,553,994; GRCh38‐v1.7), ranking among the top 0.1% most deleterious variants in the human genome, highlighting its potential functional and clinical impact.
Protein stability was evaluated using three complementary computational tools. DynaMut2, MUpro and DDGun. Both DDGun and DynaMut2 predicted destabilising effect on the tertiary structure of LDLRAP1 with a ΔΔG values of −2.5 and −1.56 kcal/mol, respectively. MUpro corroborated this conclusion with a ΔΔG of −0.95 kcal/mol, suggesting that the p.Gly54Asp substitution destabilises the protein structure.
Molecular modelling and three‐dimensional visualisation using DynaMut2 (Figure 4B) revealed significant alterations in the local intra‐protein interactions caused by the p.Gly54Asp variant. In the wild‐type protein, Gly54 forms stabilising hydrogen bonds with Ile73 and Ala144. In contrast, the Asp54 mutant establishes a new hydrogen bond with His143, a hydrophobic interaction with Ile73, and a van der waals contact with Tyr51, leading to a conformational rearrangement likely to impair proper folding and function. The Blosum substitution score for Gly → Asp (−8.563) indicates a highly non‐conservative change that is rarely tolerated in evolution. Additionally, the Kyte‐Doolittle hypopathy score (0.328) suggests that the introduction of a polar aspartate residue in a mildly hydrophobic environment is structurally unfavourable. Comparative sequence analysis across species further supports this interpretation: Gly54 is conserved in 86.4% of orthologous sequences, whereas Asp54 occurs in only 0.5%, highlighting its rarity and likely pathogenicity.
Further structural analysis using PyMOL demonstrated that the p.Gly54Asp substitution induces a significant reorganisation of the LDLRAP1–LDLR interface. In the wild‐type complex, a network of hydrogen bonds and polar interactions—(1) Ser116 with Asn^−3^, (2) Ala120 with Asn^−6^ and Ile^−7^, (3) Gln133 with Gln^+1^, (4) Trp171 with Lys^+2^, (5) Glu6 with Ser^−8^ and (6) Ile115 and Ile112 with Asn^−3^—anchors LDLRAP1 to the FxNPxY^0^ motif of LDLR. In the p.Gly54Asp mutant, these stabilising interactions are disrupted: some are weakened or shortened, while others are reoriented or more distant (Figure 5A), resulting in a reconfigured binding surface likely less effective at mediating receptor internalisation.
Structural impact of the p.Gly54Asp mutation on LDLRAP1 binding to the LDLR FxNPxY0 motif. (A) Structural modelling of the LDLRAP1–LDLR complex shows that in the wild‐type form, multiple hydrogen bonds and polar interactions stabilise the association between LDLRAP1 (light blue) and the LDLR FxNPxY0 motif (yellow). Key interactions include: (1) Ser116‐Asn−3, (2) Ala120 Asn−6/Ile−7, (3) Gln133‐Gln+1, (4) Trp171‐Lys+2, (5) Glu6‐Ser−8 and (6) Ile115/Ile112‐Asn−3. In the p.Gly54Asp variant, several of these contacts are weakened, shortened or reoriented, leading to a loss of stabilising interactions and suggesting impaired LDLR internalisation. (B) Secondary structure analysis reveals that wild‐type LDLRAP1 contains two well‐defined α‐helices (α1 and α2), whereas the p.Gly54Asp mutant displays an additional α‐helix formed by residues Ile112, Tyr113 and Arg114 (highlighted in red), indicating altered local folding and potential changes in flexibility. (C) Comparative modelling further shows that wild‐type LDLRAP1 contains eight β‐strands. Upon LDLR binding, an additional N‐terminal β‐strand from the LDLR tail aligns with LDLRAP1's β5 strand to form an extended β‐sheet. This β‐strand interaction is absent in the p.Gly54Asp mutant, suggesting reduced structural integrity and weakened receptor binding. Overall, these conformational alterations likely decrease the binding affinity between LDLRAP1 and LDLR, thereby compromising receptor‐mediated endocytosis.
Secondary structure comparison revealed conformational rearrangements, with the wild‐type LDLRAP1 comprising two α‐helices and eight β‐strands, whereas the mutant adopts three α‐helices and seven β‐strands (Figure 5B,C), suggesting local misfolding that could impair endocytic function. NetSurfP‐2.0 analysis indicated that the Gly54 is located within a β‐sheet, exposed to solvent at a relative solvent accessibility (RSA) of 18%, and lies in a structurally ordered region, making it a conformationally constrained and functionally significant site. AlphaFold predicted this region with high confidence (pLDDT > 90), and AlphaMissense classified the variant as probably pathogenic, reinforcing the predicted structural and functional disruption.
Evolutionary conservation analysis using Consurf revealed that G54 is highly conserved across species, present in 99% of aligned orthologous sequences (Profile F[G] = 99%, F[D] = 0%), underscoring its critical functional importance. Alterations at such conserved positions are often deleterious. Finally, the meta‐predictor Meta‐SNP, which integrates PANTHER, PhD‐SNP, SNAP and SIFT consistently classified the Gly54Asp variant as disease‐associated (PANTHER (0.825), PhD‐SNP (0.865), and SNAP (0.842) and a SIFT score of 0.010), with a Reliability Index of 7/10 and conservation score 0.774.
Collectively, these convergent in silico results demonstrate that the LDLRAP1 p.Gly54Asp variant destabilises protein structure, alters LDLR binding, and likely impairs receptor‐mediated endocytosis, supporting its classification as ‘likely pathogenic’ under ACMG criteria.
Discussion
4
In this study, we investigated the genetic causes of the familial hypercholesterolemia (FH) in two Tunisian families next‐generation sequencing (NGS) technology, specifically whole‐exome sequencing (WES). In the first family (Family A), we identified a previously reported splice‐site mutation, c.1845+1G>A, in the LDLR gene (19p13.2). This variant was present in both affected parents and one daughter in the heterozygous state, and in another daughter in the homozygous state, indicating an autosomal dominant pattern of inheritance (ADH) with one homozygous case. In the second family (Family B), we identified a novel homozygous mutation, c.161G>A (p.Gly54Asp), in the LDLRAP1 gene (1p36.11) in two affected brothers. According to the ACMG guidelines, the LDLRAP1 c.161G>A (p.Gly54Asp) variant meets criteria PM1, PM2, PP1 and PP3, and is therefore classified as likely pathogenic. Consistent with this classification, multiple in silico prediction tools also support a deleterious effect on protein structure and function [30].
The c.1845+1G>A splice variant, located at the highly conserved GT dinucleotide of the 5′ donor site in intron 12 of LDLR, has been previously reported in a patient from New Zealand and in two Tunisian families [27, 38] (Table 1). This variant disrupts normal mRNA splicing, resulting aberrant transcripts and reducing LDL receptor activity to < 2% in homozygous individuals. Functionally, the mutation affects the second domain of the LDLR, homologous to the epidermal growth factor (EGF) precursor. Although this domain is not essential for receptor transport or maturation, its disruption impairs acid‐dependent lipoprotein dissociation in endosomes, blocks receptor recycling, and alters the conformational structure required for efficient apoB binding [27].
Plasma lipid levels, expressed as multiples of the median (MoM), showed that the homozygous patient IV‐3 had a total cholesterol MoM of 6.31 and an LDL‐C MoM of 8.24, over threefold higher than previously reported in Jelassi et al. homozygous patients [27]. MoM values were calculated based on the lipid levels presented in Table 2. This discrepancy could be attributed to several factors, including diet, lifestyle, treatment adherence and genetic background. In our study, all patients from Family A were treated with statin (atorvastatin and rosuvastatin), and the father (III‐10) who present cardiovascular complications receives an additional cholesterol‐lowering agent that reduces intestinal cholesterol absorption (ezetimibe). No information regarding the treatment was reported in the study of Jelassi et al.
A recent meta‐analysis indicated that, although statins are commonly prescribed, individual responses vary markedly (10%–50% differences in efficacy and toxicity), influenced not only by clinical factors but also by genetic polymorphisms. In particular, the meta‐analysis highlighted that polymorphisms in SLCO1B1 play a major role in modulating the response to statin therapy. The study also mentioned other genetic variants, including APOE, LPA, SORT1 and ABCG2, which have been reported in the literature as potentially influencing statin effectiveness [39, 40]. None of these polymorphisms were identified in our patients. Jelassi et al. sequenced only LDLR and PCSK9, without broader next‐generation sequencing (NGS), which may partly explain the limited genetic insights. This limitation is further illustrated by the identification of two PCSK9 variants (p.Phe515Leu and p.Gly670Glu (rs505151)) in addition to the mutation in LDLR.
The p.Phe515Leu variant, first reported by Kotowski et al., it is extremely rare and initially classified as a variant of uncertain significance. It affects a highly conserved residue in the C‐terminal domain of PCSK9. Functional and structural analyses, particularly by Sarkar et al., showed that this variant reduces PCSK9's binding to LDL particles, enhancing LDL receptor degradation and producing an autosomal dominant FH phenotype [41, 42].
The rs505151 variant is generally considered a gain‐of‐function pathogenic variant that promotes LDLR degradation and increases plasma LDL‐C levels. Carriers of the G allele in Tunisian, Chinese and European cohorts, exhibited elevated LDL‐C, polygenic hypercholesterolemia in men, and more severe coronary atherosclerosis [43, 44, 45, 46]. In Jelassi et al.'s study, all homozygotes presented skin xanthomas, with 75% showing coronary heart diseases, whereas heterozygotes were largely unaffected [27]. In contrast, no significant association with coronary artery disease was reported in Taiwanese and Indian studies and in some cases, carriers even displayed lower LDL‐C levels [47, 48]. These observations collectively suggest that lipid profiles and clinical outcomes are multifactorial, shaped by genetic background, treatment, and environmental influences, highlighting the important considering both variant‐specific and individual context in interpreting phenotypes.
Variants in the LDLRAP1 gene, which are transmitted in an autosomal recessive manner, have not previously been reported in Tunisian patients with FH. To date, 158 ARH patients have been described worldwide, mostly from Sardinia (Italy), suggesting a founder effect. Other cases have been reported in Spain, United States, Japan, Iran, India, United Kingdom, Turkey, Mexico, Syria and Lebanon [49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68].
According to the Varsome database (https://varsome.com/gene/hg38/LDLRAP1), 454 clinical variants have been reported in the LDLRAP1. Among these, 71 are classified as pathogenic, including only six missense mutations. Additionally, 173 variants are of uncertain significance, and 216 are classified as benign. The most frequent pathogenic variant is: chr1:g.25,563,142C>A (GRCh38) (c.605C>A, p.S202Y, NM_015627.3) with an African frequency = 0.003176; https://varsome.com/gene/hg38/LDLRAP1.
The missense variant c.161G>A, (p.Gly54Asp) identified in our study lies within the PTB domain, which forms part of the lipid‐binding adaptor protein involved in multiple physiological processes, including binding to the NPXY internalisation signal of the LDLR [69].
In ARH patients, LDLR function is severely impaired in hepatocytes, despite increased receptor expression on lymphocytes, which does not result in effective LDL uptake. Fibroblasts retain partial LDL internalisation and degradation (≈70%–75% and 60%–65% of normal), distinguishing ARH from HoFH [70]. ARH lymphocytes show high surface LDLR but reduced LDL uptake, whereas LDLR‐mutant cells display more severe impairment [8]. Nevertheless, ARH cells partly downregulate LDLR in response to extracellular LDL, indicating that LDLRAP1 is essential but not the sole regulator of LDL internalisation [71]. Restoration of wild‐type LDLRAP1 cDNA rescues internalisation, confirming its critical role [72].
The LDLRAP1 variant c.161G>A (p.Gly54Asp) was classified as likely pathogenic based on its rarity, homozygous state, location within a critical functional domain, co‐segregation with affected family members, and supportive in silico prediction.
Clinically, while heterozygous parents were completely asymptomatic, the two ARH patients presented yellowish xanthomas from age 4 on knees, tendons and elbows, including a large xanthoma on the left elbow (Figure 2). LDL‐C levels were markedly elevated (5.75 and 6.90 mmol/L). These patients also exhibited a quadricuspid aortic valve (QAV); although no direct link with hypercholesterolemia has been established, it has been suggested that elevated cholesterol may contribute to valvular disease [73, 74, 75]. Screening of major valvulogenesis genes (NKX2‐5, TBX5, GATA4, MMP2) revealed no co‐segregating pathogenic variants in these ARH patients.
Variants in other valvulogenesis genes including NOTCH1, AGTR1, NKX2‐6 and BMP2 were identified in the two patients and their mother. However, their contribution remains uncertain due to lack of echocardiographic confirmation and absence of validation by Sanger sequencing. A co‐segregating synonymous variant in MMP2 was also identified. Although this variant does not alter the protein sequence, it may influence gene expression through epigenetic mechanisms, such as DNA methylation, microRNA regulation, or mRNA translation efficiency. Supporting this hypothesis, Martin et al. reported several synonymous variants co‐segregating in extended families, consistent with the inheritance patterns observed in bicuspid aortic valve (BAV) [76, 77].
It is also important to note that QAV is an extremely rare congenital anomaly in FH, with only one case previously associated with a SALL4 variant [78, 79]. Excess cholesterol during embryogenesis can disrupt Notch1 signalling, impairing valve morphogenesis through abnormal differentiation of endothelial and fibroblast cells, oxidative stress, and premature calcification. In the context of LDLRAP1 deficiency, intrauterine LDL accumulation may thus create an adverse developmental environment, indirectly affecting valve formation, highlighting the need for further research.
Collectively, these findings indicate that genetic testing is essential for the accurate diagnosis of familial hypercholesterolemia (FH), particularly in atypical cases, as it enables precise subtype classification, cascade screening, and timely intervention. By distinguishing heterozygous from homozygous or compound heterozygous patients, genetic analysis directly informs prognosis and therapeutic decisions, supporting personalised management strategies. This is especially important for rare forms such as ARH, as highlighted in our study. Early diagnosis, particularly in children, opens a critical window for preventive lifestyle measures and pharmacological therapy, including early statin initiation. Overall, integrating genetic screening with individualised treatment plans is key to reducing cardiovascular morbidity and mortality in FH, while empowering families and clinicians to implement proactive and personalised measures for cardiovascular risk prevention.
Conclusion
5
This study provides the first molecular evidence in Tunisia linking pathogenic variants in LDLR (c.1845+1G>A) and LDLRAP1 (c.161G>A; p.Gly54Asp) to familial hypercholesterolemia (FH). These findings expand the mutational spectrum of FH in North Africa and illustrate the coexistence of both autosomal dominant and recessive inheritance patterns underlying severe hypercholesterolemia. The identified variants explain the pronounced clinical phenotype observed in affected individuals and underscore the importance of integrating comprehensive genetic testing into FH diagnosis. Early molecular screening can facilitate accurate risk stratification and family counselling, while supporting precision‐based therapeutic strategies to prevent premature cardiovascular disease.
Author Contributions
W.B.N. conducted comprehensive data and bioinformatics analysis and contributed to the writing of the original manuscript. A.B.‐M. performed exome sequencing of the patients, data filtration, analysis and interpretation. H.F. oversaw the accurate collection of DNA samples and associated clinical information. F.H.K. and N.M. played a key role in patient recruitment and the collection of clinical data. H.‐G.K. critically reviewed, revised, and finalised the manuscript, providing scientific guidance and oversight throughout the study. M.M. contributed to patient recruitment and coordination of field data collection. J.‐J.H. revised the manuscript and providing scientific guidance throughout the study. L.A.K. coordinated the overall planning of the project and actively participated in drafting and structuring the manuscript. F.A. participated in drafting and structuring the manuscript.
Funding
The laboratory of J.‐J.H. is funded by internal grant QB35 at by Qatar Biomedical Research Institute, Hamad Bin Khlifa University. This work was supported by the Laboratory of Human Molecular Genetics at the Faculty of Medicine of Sfax, and the Endocrinology Department of Hedi Chaker University Hospital, Sfax.
Disclosure
Web resources: gnomAD, https://gnomad.broadinstitute.org/; Varsome, https://varsome.com/; SIFT, https://sift.bii.a‐star.edu.sg/; PolyPhen‐2, https://bio.tools/polyphen‐2; ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/; UniProt, https://www.uniprot.org/; RCSB PDB, https://www.rcsb.org/; Ensembl, https://www.ensembl.org/index.html; DynaMut2, https://biosig.lab.uq.edu.au/dynamut2/; MUpro, https://mupro.proteomics.ics.uci.edu/; DDGun, https://folding.biofold.org/ddgun/; NetSurfP‐2.0, https://services.healthtech.dtu.dk/services/NetSurfP‐2.0/; AlphaFold, https://alphafold.ebi.ac.uk/; Consurf, https://consurf.tau.ac.il/consurf_index.php; MutationTaster, https://www.mutationtaster.org/; Meta‐SNP, https://snps.biofold.org/meta‐snp/; CADD, https://cadd.gs.washington.edu/; MutPred2, https://mutpred.mutdb.org.
Consent
Informed consent for the publication of the anonymized data was obtained from all participants or their legal guardians, in accordance with ethical guidelines and the study protocol approved by the institutional review board of the Faculty of Medicine of Sfax.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Table S1: Primer sequences and corresponding annealing temperature.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1B. A. Ference , H. N. Ginsberg , I. Graham , et al., “Low‐Density Lipoproteins Cause Atherosclerotic Cardiovascular Disease. 1. Evidence From Genetic, Epidemiologic, and Clinical Studies. A Consensus Statement From the European Atherosclerosis Society Consensus Panel,” European Heart Journal 38, no. 32 (2017): 2459–2472.28444290 10.1093/eurheartj/ehx 144PMC 5837225 · doi ↗ · pubmed ↗
- 2P. Hu , K. I. Dharmayat , C. A. T. Stevens , and R. S. Jones , “Prevalence of Familial Hypercholesterolemia Among the General Population and Patients With Atherosclerotic Cardiovascular Disease: A Systematic Review and Meta‐Analysis,” (2020), accessed September 21, 2025, https://pubmed.ncbi.nlm.nih.gov/32468833/.10.1161/CIRCULATIONAHA.119.04479532468833 · doi ↗ · pubmed ↗
- 3J. P. S. Sawhney and K. Madan , “Familial Hypercholesterolemia,” Indian Heart Journal 76 Suppl 1, no. Suppl 1 (2024): S 108–S 112.38599725 10.1016/j.ihj.2023.12.002PMC 11019323 · doi ↗ · pubmed ↗
- 4L. D'Erasmo , A. Di Costanzo , and M. Arca , “Autosomal Recessive Hypercholesterolemia: Update for 2020,” Current Opinion in Lipidology 31, no. 2 (2020): 56–61.32011344 10.1097/MOL.0000000000000664 · doi ↗ · pubmed ↗
- 5R. Henderson , M. O'Kane , and V. Mc Gilligan , “The Genetics and Screening of Familial Hypercholesterolaemia,” (2016), accessed September 21, 2025, https://pubmed.ncbi.nlm.nih.gov/27084339/.10.1186/s 12929-016-0256-1PMC 483393027084339 · doi ↗ · pubmed ↗
- 6N. Ahangari , A. Sahebkar , M. Azimi‐Nezhad , et al., “A Novel Splice Site Variant in the LDLRAP 1 Gene Causes Familial Hypercholesterolemia,” Iranian Biomedical Journal 25, no. 5 (2021): 374–379.34425670 10.52547/ibj.25.5.374PMC 8487678 · doi ↗ · pubmed ↗
- 7A. J. Berberich and R. A. Hegele , “The Complex Molecular Genetics of Familial Hypercholesterolaemia,” Nature Reviews. Cardiology 16, no. 1 (2019): 9–20.29973710 10.1038/s 41569-018-0052-6 · doi ↗ · pubmed ↗
- 8C. Rodríguez‐Jiménez , D. Gómez‐Coronado , M. Frías Vargas , et al., “A New Variant (c.1A>G) in LDLRAP 1 Causing Autosomal Recessive Hypercholesterolemia: Characterization of the Defect and Response to PCSK 9 Inhibition,” Atherosclerosis 284 (2019): 223–229.30777337 10.1016/j.atherosclerosis.2019.01.010 · doi ↗ · pubmed ↗