Primary Bilateral Macronodular Adrenal Hyperplasia Associated With ARMC5 Variant and Pituitary Microadenoma
Lucía O’Connor-Ramiro, Pablo J Fernández, Julia Maroto, Ana Patiño-García, Javier Escalada, Carolina M Perdomo

TL;DR
A 63-year-old man with a rare adrenal condition and a genetic variant also had a pituitary tumor, highlighting a possible link between adrenal and pituitary issues.
Contribution
This is the second reported case linking PBMAH, an ARMC5 variant, and a pituitary microprolactinoma.
Findings
The patient had PBMAH with an ARMC5 variant and features of mild Cushing syndrome.
A pituitary microprolactinoma was identified alongside adrenal hyperplasia.
Right adrenalectomy was performed due to increased adrenal uptake on scintigraphy.
Abstract
We report a case of primary bilateral macronodular adrenal hyperplasia (PBMAH) in a 63-year-old man with a novel germline armadillo repeat-containing protein 5 (ARMC5) variant of uncertain significance (c.2525T > C; p.Phe842Ser). Imaging and clinical findings revealed markedly enlarged bilateral adrenal glands and features of mild Cushing syndrome (CS). Clinical suspicion and recommendations from guidelines prompted genetic testing. Initial management focused on controlling comorbidities and monitoring hypercortisolism. Aberrant receptor testing was negative. Progression to overt CS prompted a nor-cholesterol scintigraphy scan, revealing higher uptake in the right adrenal gland. Right adrenalectomy was performed. Concurrent findings of hypogonadotropic hypogonadism and hyperprolactinemia led to the diagnosis of a pituitary microprolactinoma on magnetic resonance imaging. To our…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
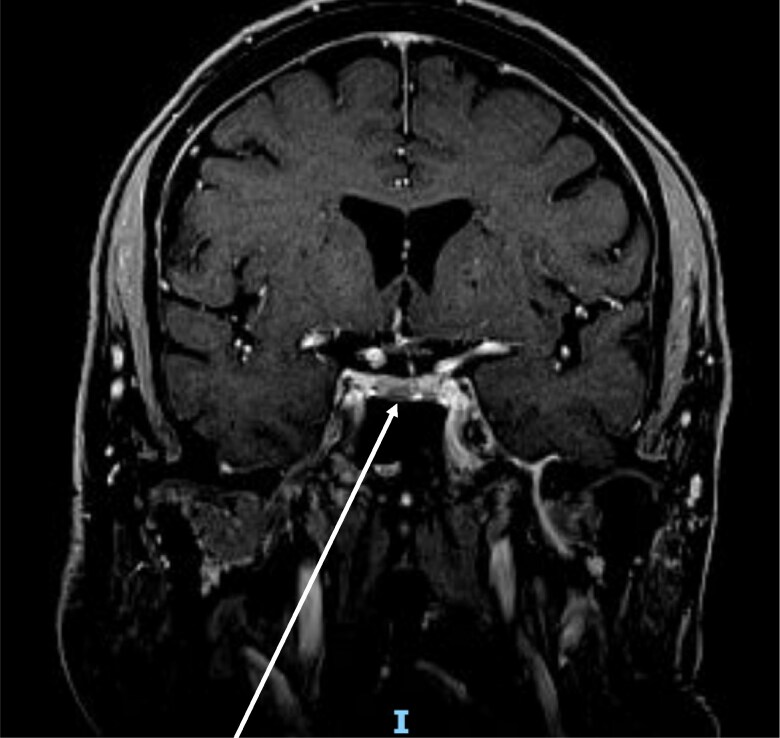 Figure 1
Figure 1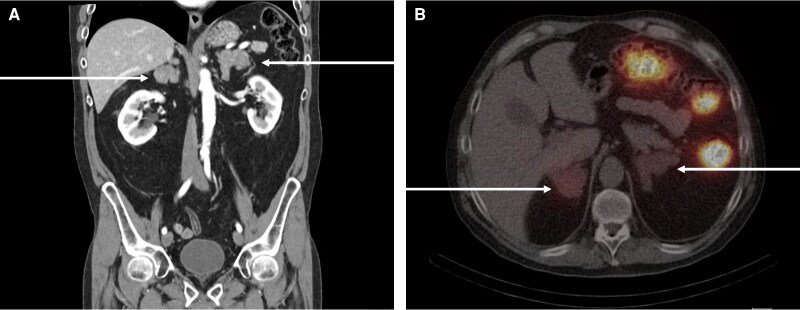 Figure 2
Figure 2| Evaluation date | Free urinary cortisol | Basal cortisol | Basal prolactin | Gonadotropins | Free testosterone |
|---|---|---|---|---|---|
| December 2021 | 77.9 µg/24 h | 11.4 µg/dL | |||
| December 2022 | 154.8 µg/24h | 11.8 µg/dL | |||
| September 2023 | 139.9 µg/24h | 15.6 µg/dL | 81.11 ng/mL | LH: 2.4 IU/L | 1.2 pg/mL |
| October 2023 | Cabergoline treatment | ||||
| February 2024 | 241.6 µg/24 h | 14.8 µg/dL | 4.57 ng/mL | 5.1 pg/mL | |
| June 2024 | 444.8 µg/24 h | 19.4 µg/dL | 2,7 ng/mL | ||
| August 2024 | Right adrenalectomy | ||||
| September 2024 | 245.5 µg/24 h | 5.2 µg/dL | 3.4 ng/mL | ||
| January 2025 | 24.8 µg/24 h | 6.32 µg/dL | 3.3 ng/mL | ||
| September 2025 | 61.7 µg/24 h | 10.68 µg/dL | 3.3 ng/mL | 6 pg/mL | |
| Date and stimuli | Nov. 15, 2023 8:43 h | Nov. 15, 2023 11:30 h | Nov. 16, 2023 8:50 h | Nov. 17, 2023 8:46 h |
|---|---|---|---|---|
| Basal cortisol | 12.2 µg/dL | 12.12 µg/dL | 13.47 µg/dL | 11.95 µg/dL |
| Cortisol at 30 min | 13.14 µg/dL | 12.16 µg/dL | 12.96 µg/dL | 11.96 µg/dL |
| Cortisol at 60 min | 14 µg/dL | 11.43 µg/dL | 13.05 µg/dL | 11.91 µg/dL |
| Cortisol at 90 min | 14.55 µg/dL | 10.94 µg/dL | 13.07 µg/dL | 11.74 µg/dL |
| Cortisol at 120 min | 14.97 µg/dL | 10.75 µg/dL | 12.57 µg/dL | 11.89 µg/dL |
| Aspect | Key features | References |
|---|---|---|
| Disease definition | Bilateral enlargement of adrenal cortex with multiple macronodules producing cortisol autonomously | [ |
| Epidemiology | Rare cause of adrenal CS; usually in middle-aged adults | [ |
| Etiology | Multifactorial: aberrant expression of G protein–coupled and other hormone receptors in adrenal nodules causing cAMP/PKA activation (eg, vasopressin, LH/hCG, GIP, β-adrenergic, angiotensin); focal/paracrine intra-adrenal production of ACTH by steroidogenic cells; somatic and germline genetic alterations (notably ARMC5) acting as drivers in a subset. | [ |
| Genetic basis | Predominantly sporadic; familial forms also reported | [ |
| ARMC5 phenotype | Larger adrenal glands; more frequent CS; occasional association with extra-adrenal tumors (eg, meningiomas) | [ |
| Genotyping indication | Bilateral adrenal enlargement with CS/MACS; family history; first-degree family screening | [ |
| Management | Unilateral adrenalectomy preferred; bilateral only if refractory. Steroidogenesis inhibitors or glucocorticoid receptor antagonists may be used. Aberrant receptor blockade occasionally effective. Lifelong follow-up required | [ |
| Prognosis | Progressive disease; morbidity from cortisol excess; improved outcomes with early detection and surgery | [ |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPituitary Gland Disorders and Treatments · Adrenal and Paraganglionic Tumors · Genetic and Clinical Aspects of Sex Determination and Chromosomal Abnormalities
Introduction
Primary bilateral macronodular adrenal hyperplasia (PBMAH) is characterized by multiple nonpigmented macronodules (>1 cm) [1]. Its morphological and clinical presentation is heterogeneous [2], and in some cases leads to cortisol overproduction and Cushing syndrome (CS). It accounts for less than 2% of endogenous CS and approximately 10% to 15% of adrenal CS cases [3]. Patients commonly present with mild autonomous cortisol secretion (MACS), which may progress to CS over time. PBMAH may be sporadic or caused by genetic mutations that promote abnormal adrenal growth [1], thereby affecting hormone regulation pathways. Germline armadillo repeat-containing protein 5 (ARMC5) mutations are a frequent genetic defect [1], responsible for most familial cases [3]. Their prevalence may reach 55% in overt CS and approximately 11% in mild CS [1].
Case Presentation
A 63-year-old White man with a history of ulcerative colitis, depression, coxarthrosis, and previously well-controlled hypertension (off medication) was referred to our endocrinology clinic in September 2023 for a second opinion after a PBMAH diagnosis at another center. Before his first evaluation at our center, the patient had high 24-hour urinary cortisol levels (154.8 μg/24 hours [Système International (SI): 427 nmol/24 hours]) (reference range: <100 μg/24 hours [SI: <276 nmol/24 hours]; assessment performed by immunoassay), with suppressed adrenocorticotropic (ACTH) levels (<5 pg/mL [SI: 1.1 pmol/L]) (reference range, 10-60 pg/mL [SI: 2.2-13.2 pmol/L]). An abdominal magnetic resonance imaging (MRI) scan from December 2022 revealed bilateral adrenal enlargement (right: 45 × 43 × 60 mm; left: 50 × 30 × 62 mm).
Diagnostic Assessment
In September 2023, on first evaluation at our center, the patient had a body mass index of 28.1, a waist circumference of 104 cm, and a blood pressure of 150/99 mm Hg; however, no typical signs of CS were evidenced. Family history was negative for adrenal disease. Laboratory analysis revealed endogenous hypercortisolism: late-night plasma cortisol of 11.45 µg/dL (SI: 316 nmol/L) (reference range, <5 µg/dL [SI: <138 nmol/L]; cortisol after 1-mg dexamethasone suppression test of 12 µg/dL (SI: 331 nmol/L) (reference range, <1.8 µg/dL [SI: <50 nmol/L] with suppressed ACTH. Furthermore, hypertension (nondipper pattern), osteoporosis, dyslipidemia, and prediabetes were diagnosed. In addition, suppressed renin levels were evidenced, with an aldosterone/renin ratio of 188. Metanephrines and catecholamines in 24-hour urine and in plasma were normal. Table 1 summarizes the hormonal assessment since the diagnosis of PBMAH.
Simultaneously, in the context of low libido, hypogonadotropic hypogonadism and hyperprolactinemia (plasma prolactin: 81.11 ng/mL [SI: 3540 pmol/L]) (reference range, 2-18 ng/mL [SI: 87-780 pmol/L]) were diagnosed. Other pituitary hormones were normal. Pituitary MRI revealed a T2-hypointense pituitary microadenoma (8 × 4 mm) (Fig. 1).
Gadolinium-enhanced pituitary magnetic resonance imaging (coronal view; T2).
Cardiovascular risk factors were controlled with medication. Osteoporosis was attributed both to hypogonadism and hypercortisolism; thus, treatment was recommended. Given mild hypercortisolism, we tested for aberrant adrenal receptors, limiting stimulation tests to those with available targeted treatment; no aberrant receptors were detected (Table 2). During follow-up in October 2023, a computed tomography scan revealed an increase in adrenal size (right: 43 × 42 × 83 mm; left: 52 × 32 × 32 × mm). After consultation with our genetics department, a next-generation sequencing panel analyzing 9 genes (aryl hydrocarbon receptor interacting protein [AIP], adenomatous polyposis coli [APC], ARMC5, cyclin dependent kinase inhibitor 1B [CDKN1B], fumarate hydratase [FH], guanine nucleotide binding protein α stimulating [GNAS], melanocortin 2 receptor [MC2R], multiple endocrine neoplasia 1 [MEN1], and protein kinase cyclic adenosine monophosphate–activated catalytic subunit α [PRKACA] was requested. A heterozygous ARMC5 variant (c.2525T > C; p.Phe842Ser) was identified. Genetic counseling was offered.
Treatment
Pharmacological treatment for comorbidities initially included the following: spironolactone 100 mg daily (suspected primary hyperaldosteronism), pravastatin 20 mg daily, alendronic acid 70 mg weekly, vitamin D3 2000 IU (6 drops at breakfast), and cabergoline 0.5 mg (half a tablet, twice weekly). In December 2023, after the captopril challenge test, primary hyperaldosteronism was ruled out, and spironolactone was modified to losartan. In June 2024, free urinary cortisol levels exceeded 3 times the upper limit, and a nor-cholesterol scintigraphy was performed to guide unilateral adrenalectomy. The scan showed greater uptake in the right adrenal gland (Fig. 2B). Right adrenalectomy was performed in August 2024 without complications. Postoperatively, the patient developed adrenal insufficiency; basal cortisol was 5.2 µg/dL (143 nmol/L) (reference range, >10 µg/dL [SI: >275 nmol/L] with a cortisol response to ACTH stimulation of 17 µg/dL (reference range, >18 µg/dL [SI: >496 nmol/L]), thus oral hydrocortisone was indicated.
A, Computed tomography of the abdomen showing the massively enlarged bilateral adrenal glands. B, Nor-cholesterol scintigraphy revealed a greater uptake in the right adrenal gland.
Outcome and Follow-up
The patient received glucocorticoid support until March 2025, after demonstrating a basal cortisol level of 10.68 µg/dL. At his most recent medical consultation, he reported improved mood, decreased fatigue, and increased libido. He reports having enough energy to engage in at least 30 minutes of daily exercise. Hypertension treatment was suspended because ambulatory blood pressure levels were normal. A second pituitary MRI was performed in September 2025, and the disappearance of the adenoma was evidenced.
Discussion
We report a case of PBMAH with overt CS in a patient carrying a previously undescribed germline ARMC5 variant (c.2525T > C; p.Phe842Ser), classified as of uncertain significance. PBMAH is a rare adrenal disorder, though its true prevalence may be underestimated [3]. Adrenal incidentalomas occur in 1% to 5% of the population, with 2.7% to 10% being bilateral [4]. MACS is present in 35% to 40% of these bilateral incidentalomas, and some may represent PBMAH. Table 3 summarizes the key features of PBMAH.
While most PBMAH cases are sporadic, a substantial proportion involve germline ARMC5 mutations [5]. More than 265 mutations have been identified [4, 6], including several familial cases with autosomal dominant inheritance [1, 2]. ARMC5 is the primary genetic cause of PBMAH, accounting for up to 80% of familial and 30% of sporadic cases [1-3]. In an Italian cohort, 18.8% of patients with MACS carried pathogenic ARMC5 variants [7].
The c.2525T > C; p.(Phe842Ser) ARMC5 variant was initially classified as a variant of unknown clinical significance according to the American College of Medical Genetics and Genomics [8] on fulfilment of the PM2 (“absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium”) and PP4 (“patient's phenotype or family history is highly specific for a disease with a single genetic etiology”). To definitively relate the presence of this variant with disease, segregation analysis of family members will be performed for fulfillment of PS2 and/or PP1 criteria, which would lead to final classification of the variant as “likely pathogenic.”
Of particular interest in this case is the co-occurrence of a prolactin-secreting pituitary microadenoma. This is the second case report of a patient with both PBMAH and a pituitary adenoma, presenting with a germline ARMC5 variant [9]. ARMC5 is believed to act as a tumor suppressor gene, inhibiting abnormal cell proliferation [5]. It encodes a protein predominantly located in the cytoplasm, whose function depends on interactions with other proteins [5]. Loss of ARMC5 function may contribute to tumor formation through a 2-hit mechanism—a germline mutation followed by a somatic event that affects the second allele.
Inactivation of ARMC5 in the adrenal cortex is associated with progressive dedifferentiation of adrenocortical cells, growth of bilateral masses, and inefficient steroidogenesis [6]. Beyond PBMAH, ARMC5 mutations have been associated with other neoplasms, including intracranial meningiomas [1, 2]. Its high expression in the pituitary gland [10] suggests a potential, though unconfirmed, role in pituitary tumorigenesis. It remains unknown whether this specific variant predisposes to microadenoma development. Other endocrine comorbidities, such as primary hyperaldosteronism, hyperparathyroidism, and multinodular goiter, have also been described in carriers of ARMC5 pathogenic variants [11]. Likewise, associations with breast, thyroid, and parathyroid cancer have been reported [11, 12], although more studies are needed to clarify these links.
Other genetic causes of PBMAH include inactivation of lysine demethylase type 1A (KDM1A), which has been linked to hereditary food-dependent CS (glucose-dependent insulinotropic peptide–dependent PBMAH) [13]. This mutation is also associated with adrenal myelolipoma, monoclonal gammopathy of undetermined significance, and multiple myeloma. PBMAH may also result from alterations in MC2R, PRKACA, and phosphodiesterase (PDE)11A genes [1, 2], and it can occur as part of familial tumor syndromes such as McCune-Albright syndrome (GNAS), MEN1, familial APC, or hereditary leiomyomatosis and renal cell carcinoma (FH gene variant). Genetic testing may aid in the earlier diagnosis and management of PBMAH [5], though there are currently no established criteria for screening. Given the delayed onset and frequent lack of symptoms, genetic screening of first-degree relatives may help identify individuals at risk [1].
Assessment of comorbidities associated with cortisol excess, such as diabetes, hypertension, and osteoporosis, is also crucial. Aberrant adrenal receptors are found in approximately 30% of cases of bilateral and unilateral adrenal hyperplasia [2] and can be identified through stimulation tests that evaluate abnormal cortisol responses [1]. Although receptor blockade rarely achieves long-term control, it may offer other treatment options [2, 14]. Testing under dexamethasone suppression may help minimize ACTH interference [1].
In patients with clinically significant hypercortisolism, surgical intervention is often necessary [1, 2]. Bilateral adrenalectomy offers definitive control but requires lifelong steroid replacement and carries substantial risk [2]. Therefore, unilateral adrenalectomy is preferred when possible, targeting the gland with a higher uptake on iodocholesterol scintigraphy of the larger gland. In our case, the glands were similar in size, and scintigraphy guided the decision. Adrenal venous sampling (AVS) is generally unhelpful in asymmetric PBMAH since the larger gland usually secretes more cortisol in AVS [15], though it may be helpful in symmetric presentations [14].
Medical therapy, such as ketoconazole or mitotane, can be used before surgery or for long-term management in patients who are not surgical candidates [1, 2]. Low-dose ketoconazole has been successfully used for up to 10 years in selected cases [16]. Receptor-targeted treatments, such as propranolol, octreotide, long-acting gonadotropin–releasing hormone (GnRH) agonists, or angiotensin receptor blockers, may be considered when appropriate [2]. However, their effectiveness varies, and they may not result in clinical improvement. Mifepristone (a glucocorticoid receptor antagonist) and osilodrostat (an adrenostatic agent) may also be considered medical options for PBMAH when surgery is not feasible [17, 18].
Lifelong follow-up is essential [19, 20], as recurrence of hypercortisolism is very likely; it occurs in 10% to 68% of cases after unilateral adrenalectomy, sometimes years later [2, 18]. In selected cases, total adrenalectomy of the larger gland, combined with partial resection of the contralateral gland, may reduce the risk of recurrence. Our patient will be evaluated every 6 to 12 months. The hypothalamic-pituitary-adrenal axis and hypercortisolism will be assessed periodically. The withdrawal of cabergoline is planned; thus, MRI revealed the disappearance of the prolactinoma after 18 months of treatment. In conclusion, this case contributes to the expanding phenotype of ARMC5-associated disease and raises questions about its role in the pathogenesis of pituitary tumors. Although causality cannot be confirmed, this association warrants further investigation.
Learning Points
PBMAH is a complex endocrine disorder with variable clinical presentations frequently associated with ARMC5 mutations. ARMC5 pathogenic variants are associated with a more severe phenotype of PBMAH.We have described a variant of uncertain significance of the ARMC5 gene in a patient with PBMAH and pituitary abnormalities. It is unknown if this variant is associated with these comorbidities.The spectrum of ARMC5-related tumors must be outlined; in the meantime, patients should be monitored for other neoplastic entities. ARMC5 screening might be recommended in patients with PBMAH and MACS or CS.
Contributors
All authors made individual contributions to the authorship. L.O., P.F., and C.P. were involved in the case presentation, diagnosis, and patient care, as well as the creation of graphs. J.M., A.P., and J.E. were involved in case diagnosis. J.E. was involved in case follow-up. All authors reviewed and approved the final draft.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Vassiliadi DA, Tsagarakis S. Diagnosis and management of primary bilateral macronodular adrenal hyperplasia. Endocr Relat Cancer. 2019;26(10):R 567‐R 581.32053747 10.1530/ERC-19-0240 · doi ↗ · pubmed ↗
- 2Araujo-Castro M, Marazuela M. Cushing's syndrome due to bilateral adrenal cortical disease: bilateral macronodular adrenal cortical disease and bilateral micronodular adrenal cortical disease. Front Endocrinol (Lausanne). 2022;13:1016728.10.3389/fendo.2022.913253 PMC 938904035992106 · doi ↗ · pubmed ↗
- 3Charchar HLS, Fragoso MCBV. An overview of the heterogeneous causes of Cushing syndrome resulting from primary macronodular adrenal hyperplasia (PMAH). J Endocr Soc. 2022;6(5):bvac 045.35402764 10.1210/jendso/bvac 041PMC 8989153 · doi ↗ · pubmed ↗
- 4Chevalier B, Vantyghem MC, Espiard S. Bilateral adrenal hyperplasia: pathogenesis and treatment. Biomedicines. 2021;9(10):1802.34680514 10.3390/biomedicines 9101397 PMC 8533142 · doi ↗ · pubmed ↗
- 5Assié G, Libé R, Espiard S, et al ARMC 5 mutations in macronodular adrenal hyperplasia with Cushing syndrome. N Engl J Med. 2013;369(22):2105‐2114.24283224 10.1056/NEJ Moa 1304603 PMC 4727443 · doi ↗ · pubmed ↗
- 6Bouys L, Chiodini I, Arlt W, et al Update on primary bilateral macronodular adrenal hyperplasia (PBMAH). Endocrine. 2021;71(3):595‐603.33587256 10.1007/s 12020-021-02645-w · doi ↗ · pubmed ↗
- 7Morelli V, Elli FM, Frigerio S, et al Prevalence and clinical features of armadillo repeat-containing 5 mutation carriers in a single-center cohort of patients with bilateral adrenal incidentalomas. Eur J Endocrinol. 2023;189(2):242‐251.37625448 10.1093/ejendo/lvad 088 · doi ↗ · pubmed ↗
- 8Richards S, Aziz N, Bale S, et al Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐424.25741868 10.1038/gim.2015.30PMC 4544753 · doi ↗ · pubmed ↗