Rational Electrolyte Structure Engineering for Highly Reversible Zinc Metal Anode in Aqueous Batteries
Yi Zhuang, Yukai Liang, Wenyao Zhang, Yuntong Sun, Zhenxing Wang, Jingyan Guan, Boyuan Zhu, Junjie Cui, Jiahao Tang, Jong-Min Lee, Junwu Zhu

TL;DR
This review explores how to design better electrolytes for zinc batteries to improve performance and safety.
Contribution
The paper provides a systematic review of electrolyte engineering strategies to enhance zinc metal anodes in aqueous batteries.
Findings
Rational electrolyte design can mitigate dendrite growth and parasitic reactions in zinc anodes.
Optimization of zinc salts and additives improves electrochemical performance.
Structure–property relationships in electrolytes are key for next-generation battery development.
Abstract
This review systematically summarizes the electrochemical principles governing Zn2+ nucleation and deposition, elucidating their intrinsic correlations.The review discusses zinc salt optimization, electrolyte additives, and novel electrolyte designs, providing mechanistic insights into anodic Zn2+ electrodeposition.The review proposes future directions for aqueous zinc metal anode, including dynamic reconstruction, AI-guided additive screening, etc. This review systematically summarizes the electrochemical principles governing Zn2+ nucleation and deposition, elucidating their intrinsic correlations. The review discusses zinc salt optimization, electrolyte additives, and novel electrolyte designs, providing mechanistic insights into anodic Zn2+ electrodeposition. The review proposes future directions for aqueous zinc metal anode, including dynamic reconstruction, AI-guided additive…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 10
Figure 10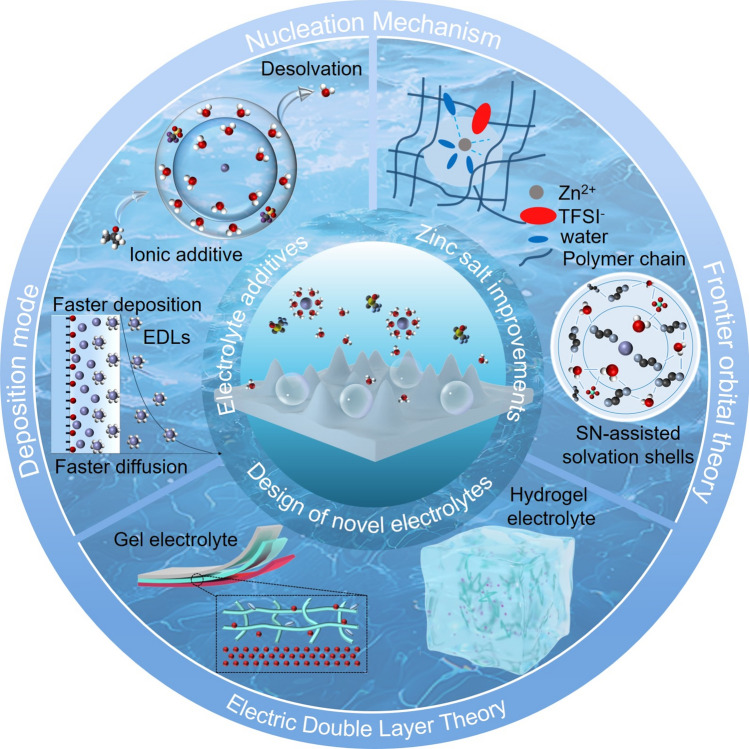 Figure 1
Figure 1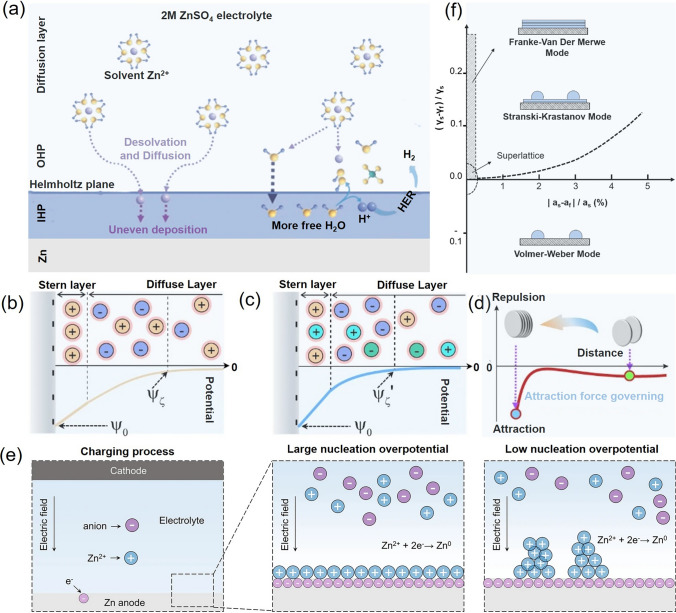 Figure 2
Figure 2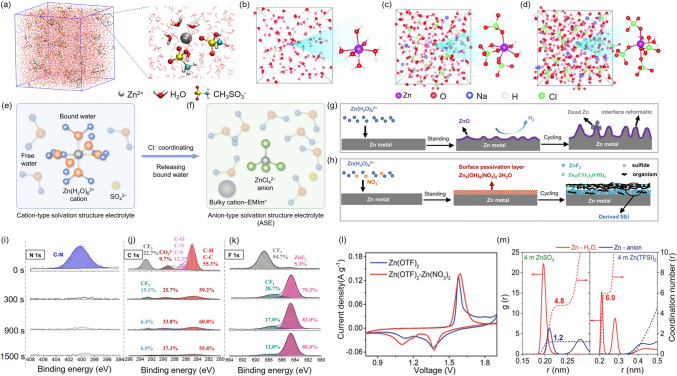 Figure 3
Figure 3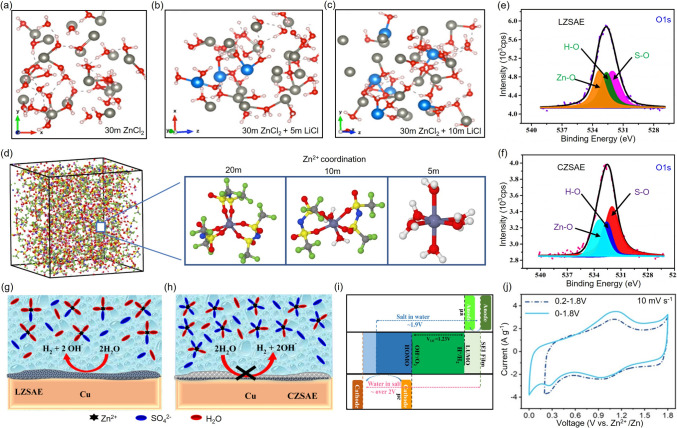 Figure 4
Figure 4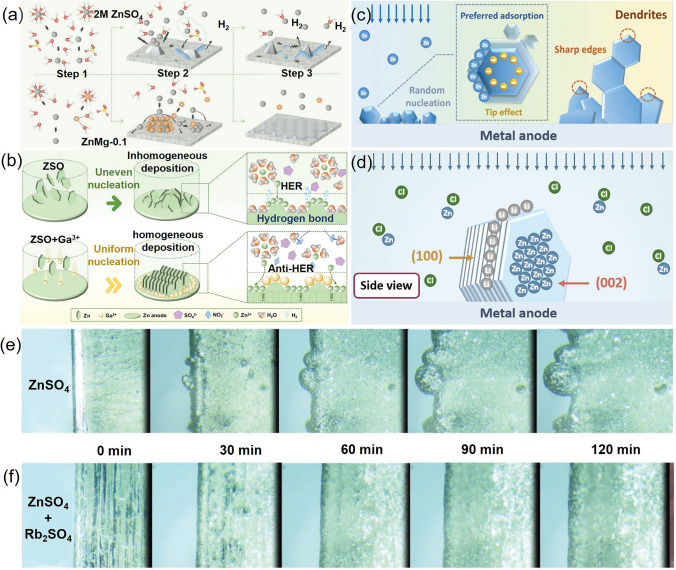 Figure 5
Figure 5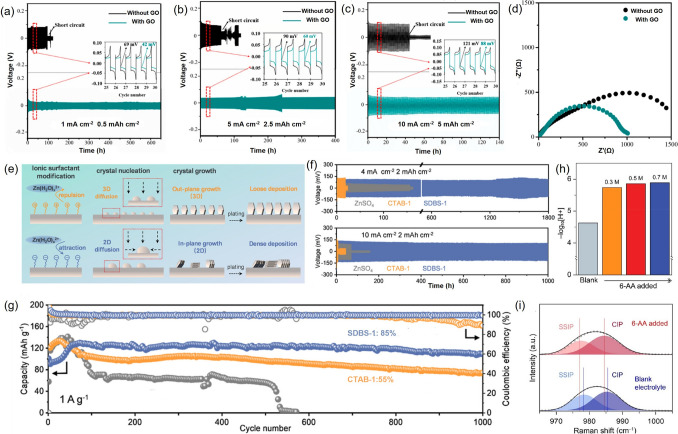 Figure 6
Figure 6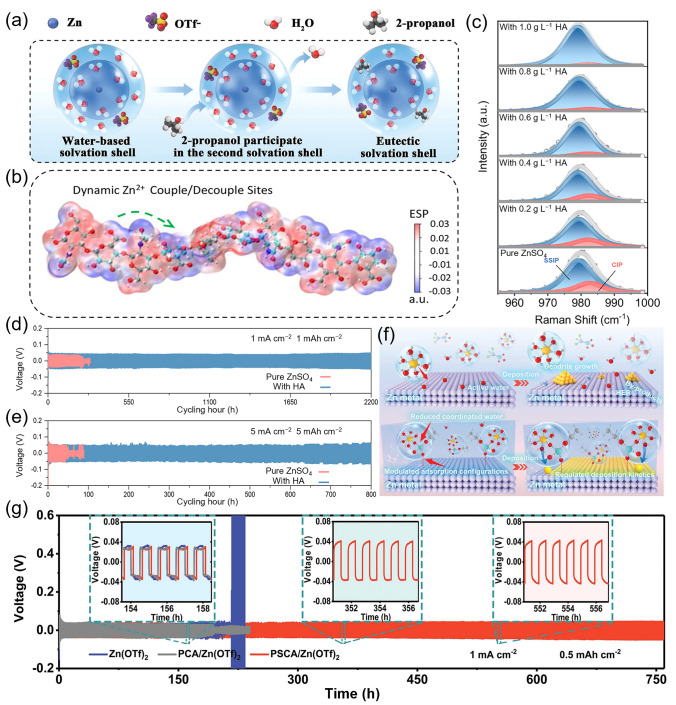 Figure 7
Figure 7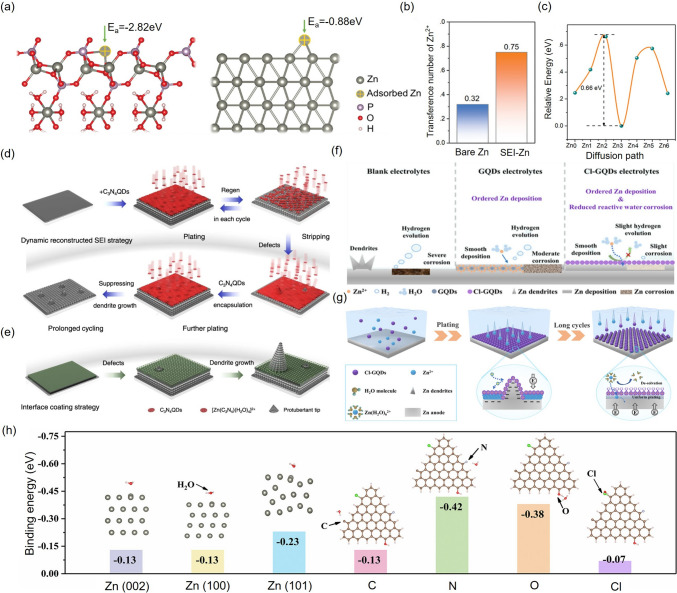 Figure 8
Figure 8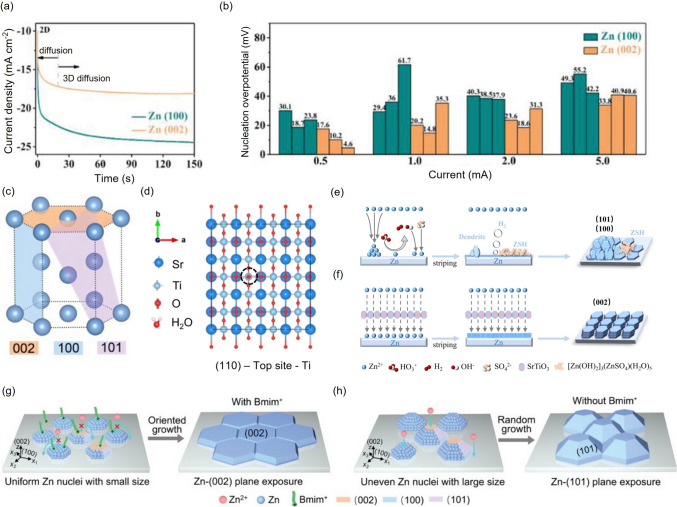 Figure 9
Figure 9 Figure 11
Figure 11Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvanced battery technologies research · Membrane-based Ion Separation Techniques · Advancements in Battery Materials
Introduction
The rapid depletion of fossil fuels and escalating environmental concerns have intensified the demand for advanced energy storage technologies, making their development a critical global priority [1]. Among sustainable energy solutions, rechargeable batteries are pivotal, providing vital energy storage and conversion for electronic devices and transportation systems, playing an indispensable role in the pursuit of carbon neutrality.
Aqueous electrolytes offer ionic conductivities that are two orders of magnitude higher than the organic counterparts and, importantly, exhibit outstanding intrinsic safety. Therefore, aqueous metal-ion batteries are emerging as high-safety candidates for large-scale energy storage applications. In particular, aqueous zinc-ion batteries (AZIBs), using zinc ions (Zn^2+^) as charge carriers, have attracted considerable attention due to their low redox potential (− 0.76 V vs. standard hydrogen electrode, SHE), high theoretical capacity (5855 mAh cm⁻^3^ and 820 mAh g⁻^1^), intrinsic safety, and facile processing [2–4]. Revisiting the Zn^2+^ storage mechanism based on Zn^2+^/Zn redox couple (Zn^2+^ + 2e^−^ ⇌ Zn), the interfacial zinc chemistry at the electrode/electrolyte interface plays a critical role, especially for the metallic Zn anode. In neutral electrolytes, solvated Zn^2+^ forms a stable hydration shell with abundant polar water molecules, while water molecules preferentially adsorb onto the Stern layer of the electric double layer (EDL), leading to parasitic water reduction and increased internal pressure. The associated hydrogen evolution reaction (HER) fluctuates the pH value in local areas, enriching anions (e.g., OH^−^, SO_4_^2−^), which subsequently undergo complexation and generate insulating and passivating zinc byproducts. These processes continuously consume active Zn^2+^ and hinder ion/electron transport [5–7]. Furthermore, the uncontrolled growth of zinc dendrites, arising from the non-uniform Zn^2+^ flux, desolvation, and nucleation barriers, remains a crucial obstacle, similar to other (alkali) metal anodes. The issues of HER, passivation, and dendrite formation are interrelated: dendrite growth enlarges the anode surface area and accelerates HER; HER elevates local OH^−^ concentration and promotes inert byproduct formation; these byproducts deposit unevenly, exacerbate electric field polarization, and further stimulate dendrite growth. These synergistic degradation leads to capacity fading, reduced Coulombic efficiency (CE), and shortened battery lifespan.
To address these challenges, tremendous efforts have been devoted to moderating the electrochemical behavior of Zn^2+^ and water molecules, including anode design [8], artificial interphase engineering [9–11], electrolyte structure regulation [12, 13], charging protocol optimization [14], etc. Among these, electrolyte engineering has emerged as a pivotal strategy for advancing Zn metal anode in AZIBs, owing to its profound influence on Zn^2+^ solvation structure, ion flux, migration dynamics, and nucleation behavior, and in situ formation of functional interphases. Early approaches focused on adjusting salt types and concentration to tailor Zn^2+^ solvation, thereby suppressing water-induced side reactions by weakening water activity. In addition to these effects, many additives promote in situ formation of protective interphases that isolate the reactive water molecules, offering ion sieving capabilities that modulate Zn^2+^ desolvation and enable uniform nucleation on the Zn anode, mitigating dendrite growth. Recently, a dynamic interphase concept [15] has been proven to achieve real-time conformal contact with the Zn anode, continuously regulating deposition behavior and enhancing long-term anode stability. Beyond this, a range of emerging concepts and theoretical frameworks have further advanced the landscape of electrolyte design.
In this review, we systematically summarize the electrochemical principles governing Zn^2^⁺ nucleation and deposition, while elucidating their intrinsic interrelationships. Adopting a chronological framework, we discuss key developments in (i) zinc salt optimization, (ii) functional electrolyte additives, and (iii) the design of novel electrolyte systems (Fig. 1), providing mechanistic insights into Zn^2^⁺ electrodeposition from the perspective of electrolyte engineering. In contrast to prior reviews that classify additives by chemical composition, we organize the additive section based on dominant functional mechanisms, including electrostatic shielding, interfacial adsorption, desolvation modulation, in situ solid electrolyte interphase (SEI) formation, and crystal-plane engineering. These mechanisms are critically evaluated to reveal the fundamental processes underlying improved electrochemical performance. Furthermore, we highlight recent advances in dynamic interfacial construction, emphasizing real-time, self-regulating stabilization strategies under cycling conditions. Finally, we summarize the recent progress, existing challenges, and provide prospects in electrolyte engineering for Zn metal anode in AZIBs, aiming to inspire new insights and accelerate their practical deployment.Fig. 1. Electrolyte structure regulation strategies for Zn metal anodes in AZIBs
Electrochemical Theory of Zn2+ Nucleation and Growth
The electrochemical performance of Zn metal anode in AZIBs is critically influenced by the nucleation particle size and nucleation energy barrier, two key parameters that dictate Zn^2+^ deposition behavior and consequently affect both cycling stability and battery lifespan. The EDL model provides an effective theoretical framework for elucidating the nucleation and growth mechanisms at the anode/electrolyte interface. In conventional ZnSO_4_ electrolytes, solvated Zn^2+^ ions, primarily in the form of [Zn(H_2_O)6]^2+^, diffuse from the diffusion layer to the outer Helmholtz plane (OHP) and undergo desolvation under long-range electrostatic interactions. The energy barrier associated with this desolvation process governs the kinetics of Zn^2+^ deposition [16]. Additionally, the potential drop across the Helmholtz layer serves as a crucial descriptor for nucleation and growth, influencing both the critical nucleation size and nucleation rate. The electric field distribution within the EDL further modulates Zn^2+^ deposition morphology [17]. Therefore, an in-depth understanding of these descriptors and interfacial parameters from an electrochemical theory perspective is essential for rationalizing Zn^2+^ nucleation behavior and guiding the design of high-performance AZIBs.
Electric Double Layer Theory
To elucidate the spatial arrangement of ions and solvent molecules at the anode/electrolyte interface, the EDL theory provides a fundamental framework for understanding interfacial reactions and performance-enhancing mechanisms in AZIBs. According to the Stern model, the EDL in AZIBs is composed of two distinct regions: the Stern layer and the diffusion layer. The Stern layer comprises the inner Helmholtz plane (IHP) and the outer Helmholtz plane (OHP) [16]. The IHP contains specifically adsorbed ions directly adjacent to the electrode surface, whereas the OHP host partially desolvated Zn^2^⁺ ions and a portion of the anions. Beyond the OHP lies the diffusion layer, extending into the bulk electrolyte. During deposition, Zn^2+^ migrates from the diffusion layer to the OHP, undergoes desolvation, and is subsequently reduced and deposited within the IHP(Fig. 2a) [18].Fig. 2a Schematic illustration of Zn deposition process [18]. Comparison of the EDL structures of Zn deposits in b ZnSO_4_ and** c** La^3+^-ZnSO_4_ electrolytes [21]. d Proposed growth models of Zn deposits in La^3+^-ZnSO_4_ electrolytes [21]. e Schematic diagram of the effect of overpotential on Zn deposition behavior [28]. f Three nucleation regions mapped as a function of lattice mismatch (x-axis) and surface energy differences (y-axis), where γ_s_ and γ_f_ represent the surface energies of the substrate and deposited film, respectively, and a_s_ an a_f_ denote their corresponding lattice parameters [23, 30]
Hydrogen evolution and by-product formation, two key factors affecting the cycling stability and long-term performance of Zn anodes, are closely linked to the EDL, whose structure and composition critically govern interfacial reactivity [16]. Taking the ZnSO_4_-based electrolyte as a representative example, hydrated Zn^2+^ ions [Zn(H_2_O)6]^2^⁺ migrate from the bulk solution into the EDL during charging. Upon partial or complete desolvation, Zn^2^⁺ ions are reduced and electrodeposited onto the anode surface. However, incompletely desolvated species may reach the interface, triggering parasitic hydrogen evolution and corrosion of the zinc anode. Additionally, the preferential accumulation of water dipoles and sulfate anions (SO_4_^2^⁻) within the EDL intensifies electrostatic repulsion among adjacent Zn nuclei, disrupting uniform nucleation and resulting in dispersed, loosely packed platelet-like deposits [19]. The extent of electrostatic repulsion is determined by the EDL thickness \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$(\delta )$$\end{document} , which, for a planar interface, can be expressed analytically as a function of the Debye length ( \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$${\kappa }^{-1}$$\end{document} ) and the dimensionless surface charge density ( \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$\sigma$$\end{document} ). The explicit form of \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$\delta$$\end{document} is given by Eq. (1) [20]:
\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$\delta =\frac{1}{\kappa }\text{ln}\left(\frac{\text{tanh}\left(\frac{{\text{sinh}}^{-1}\frac{\sigma }{2}}{2}\right)}{\text{tanh}\left(\frac{{\text{sinh}}^{-1}\frac{\sigma }{2}}{200}\right)}\right)$$\end{document}Herein, tanh⁻^1^ and sinh⁻^1^ represent the inverse hyperbolic tangent and inverse hyperbolic sine functions, respectively.
A decrease in the EDL thickness δ, achieved by reducing the Debye length \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$${\kappa }^{-1}$$\end{document} or the surface charge density \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$\sigma$$\end{document} , effectively migrates inter-platelet electrostatic repulsion and facilitates dense, uniform zinc electrodeposition. As a representative strategy, Qie et al. introduced La(NO_3_)3 into an aqueous electrolyte. The high-valent La^3^⁺ cations competitively replace Zn^2^⁺ at the interface, thereby decreasing the net surface charge of the nascent zinc layer. As shown in Fig. 2b–d, this charge attenuation leads to a significant contraction in \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$\delta$$\end{document} , resulting in improved cycling stability and enhanced electrochemical performance of the cell [21].
Nucleation Mechanism of Zn2+
Nucleation Barrier
Zinc deposition initiates with nucleation: Zn^2+^ ions first migrate toward the anode–electrolyte interface under the influence of an electric field and then nucleate on the Zn anode surface after overcoming the corresponding nucleation energy barrier [22]. A high nucleation barrier can limit the number of active nucleation sites, thereby promoting non-uniform zinc deposition (Fig. 2e). During continued deposition, Zn^2+^ ions undergo two-dimensional surface diffusion, which lead to Volmer–Weber growth characterized by island-like, inhomogeneous deposition. This uneven morphology disrupts the electric field distribution and ultimately induces the tip effect. As tip curvature increases, the local electric field intensity and surface charge density rise, enabling Zn^2+^ ions in the vicinity to overcome the nucleation barrier more readily and initiate dendritic growth. Moreover, ion concentration plays a vital role in the nucleation process: regions with higher ionic concentration or faster ion transport exhibit reduced nucleation barriers [23, 24]. Recent studies have demonstrated that both the binding energy between the Zn^2+^ and the anode substrate, and lattice adaptability significantly influence the energy barrier for nucleation.
To suppress dendrite formation, Zhao et al. introduced cyclodextrin (α-CD) as an electrolyte additive. The α-CD molecules interact strongly with metallic zinc, preferentially adsorbing onto the anode surface and lowering the nucleation barrier. This adsorption also induces additional surface charge on the zinc, generating enhanced electrostatic attraction for Zn^2+^ and guiding its deposition. Experimental results show that the nucleation overpotential on Zn foil decreases progressively with increasing α-CD concentration, confirming its effectiveness in reducing the zinc nucleation barrier. As a result, the α-CD additive promotes three-dimensional Zn^2+^ diffusion, leading to more uniform deposition [25].
Nucleation Particle Size
The nucleation particle size also plays a critical role in determining the electrochemical performance of Zn anodes. According to classical homogeneous nucleation theory, the critical radius (r) of the spherical nucleus is given by:
\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$r=2\frac{\gamma {V}_{\text{m}}}{F|\eta |}$$\end{document}where γ is the surface energy at the anode–electrolyte interface, Vm is the molar volume of Zn, F is the Faraday constant, and η is the nucleation overpotential. This expression indicates that smaller nucleation particle size corresponds to higher polarization overpotentials, which in turn enhance the nucleation driving force and promote finer, more uniform zinc deposition [18, 26]. Zhou et al. utilized ZnCl_2_-based electrolytes to investigate how electrolyte concentration influences the nucleation radius and Zn deposition behavior. In situ atomic force microscopy (AFM) observations revealed that in dilute electrolytes, large and sparsely distributed zinc nuclei were formed. In contrast, medium-concentration electrolytes minimized the γ/η ratio, leading to the formation of smaller zinc nuclei and enabling uniform, dense deposition of zinc species [27].
Nucleation Rate
The nucleation rate significantly influences the stability of the Zn anode, as a higher nucleation rate promotes more uniform Zn^2+^deposition during cycling. The nucleation rate (ω) can be described by Eq.:
\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$\omega =K\text{ exp}\left(-\frac{\pi h{\sigma }^{2}LA}{\rho nFRT\eta }\right)$$\end{document}where K is the pre-exponential factor, σ is the interfacial tension, L is Avogadro number, n is the valence number of the metal ion, R and F are the gas constant and Faraday’s constant, T is the absolute temperature, and ρ*, *h, and A are the density, atomic height, and atomic weight of the deposited metal Zn, respectively. This expression indicates that the nucleation rate increases exponentially with the increase in overpotential, thereby allowing the formation of a uniform and compact Zn layer. Leveraging this principle, Hu et al. introduced sodium L-tartrate (Na-L) into the ZnSO_4_ solution to regulate the nucleation overpotential and improve zinc deposition uniformity. With the Na-L additive, the nucleation overpotential increased from 28.3 to 45.9 mV. Chronoamperometry measurements further revealed that Na-L enabled uniform three-dimensional diffusion of Zn^2+^, as evidenced by the rapid stabilization of the current density, suggesting homogeneous crystal growth during electrodeposition [28].
Growth Mode of Deposits
The physicochemical properties of the substrate, particularly the lattice mismatch between zinc crystals and the substrate, directly affect its zincophilicity [29]. Accordingly, three classical deposition growth modes have been proposed based on the degree of lattice mismatch and interfacial energy differences between the deposit and the substrate: the Frank–Van der Merwe mode, the Volmer–Weber mode, and the Stranski–Krastanov mode (Fig. 2f) [5, 23, 30]. When the lattice mismatch is minimal, epitaxial growth favors smooth and continuous film deposition, characteristic of the Frank–Van der Merwe growth mode. As the mismatch increases, deposition behavior transitions to the Stranski–Krastanov mode, where layer-by-layer growth is followed by island formation. In cases of significant mismatch, where the interatomic forces within the deposited atoms surpass the adhesion to the substrate, the Volmer–Weber mode dominates, resulting in discrete island growth.
Based on this framework, regulating the lattice mismatch between the substrate and zinc enables controlled zinc nucleation and growth, leading to compact, dendrite-free deposits. For instance, Li et al. introduced maleic anhydride (MA) as an additive, which preferentially adsorbs on the (002) facet of Zn. This surface-selective interaction guides the deposition toward the Frank–Van der Merwe mode, thereby suppressing dendrite formation and enhancing uniformity [6]. Similarly, Ma et al. introduced sodium hyaluronate (SH) in ZnSO_4_ electrolyte. The SH exhibits a strong affinity for Zn, restructuring the electric double layer and forming a dynamic Zn-SH* interface. This promotes the preferential growth of Zn along the (002) plane, attributed to the polar functional groups in SH that improve surface adsorption. Moreover, the enhanced wettability of the SH-modified electrolyte facilitates Frank–Van der Merwe-type deposition, yielding a uniform and dense Zn deposition [31].
Frontier Orbital Theory
In the development of electrolyte additives, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels, derived from frontier molecular orbital theory, are crucial parameters for screening solvents and additives in electrolyte systems [32, 33]. A higher HOMO energy level enhances a molecule’s electron-donating ability, thereby facilitating coordination with metal ions or adsorption on electrode surfaces. In contrast, a lower LUMO energy level indicates a stronger electron-accepting ability, promoting the reduction and deposition of metal ions. Molecules that exhibit both low HOMO and high LUMO energies typically possess strong chemisorptive properties. Density functional theory (DFT) calculations of HOMO–LUMO levels are therefore widely employed to guide the rational selection of electrolyte components. The energy difference between HOMO and LUMO is closely related to both the electrochemical stability window and the electrode potential. Ideally, the electrode potential must lie within the HOMO–LUMO energy range of the electrolyte to prevent decomposition [34]. For instance, when the Fermi level of a cathode material is lower than the HOMO level of the electrolyte, electrons can transfer from the electrolyte to the cathode, leading to oxidation of the electrolyte. Hence, the working potentials of both anode and cathode must fall within the HOMO–LUMO gap of the electrolyte system to ensure stability [35, 36].
Additives with a narrow HOMO–LUMO gap can diminish interfacial charge-transfer resistance, thereby lowering the nucleation barrier for zinc deposition. For example, Wang et al. employed a system comprising Zn(BF_4_)2·4H_2_O salt and vinylene carbonate (VC) solution, where DFT calculations revealed a decreased HOMO–LUMO gap and elevated HOMO level in the solvated structure. VC exhibits a higher HOMO level at − 6.96 eV compared to water (− 8.06 eV), indicating enhanced electron-donating capability and improved charge transfer, thus increasing the reduction stability of the electrolyte [37]. Additionally, LUMO and HOMO levels are strongly associated with SEI formation. Additives with low LUMO levels preferentially accept electrons and are reduced at the electrode surface, facilitating SEI formation and uniform Zn^2^⁺ deposition [38]. Han et al. introduced tetradecafluorononane-1,9-diol (TDFND) as an additive, which, with a low LUMO of 0.10 eV significantly lower than that of water, underwent preferential reduction to form a ZnF_2_-rich SEI layer [39]. Li et al. employed sodium diethyldithiocarbamate (DDTC) and calculated the LUMO for H_2_O, Zn^2^⁺–H_2_O, DDTC⁻, and Zn–DDTC to be 2.06, − 0.75, 0.01, and − 1.88 eV, respectively. This indicates that DDTC coordinated with Zn^2⁺^ undergoes preferential reduction, contributing to SEI formation on the anode surface [40]. Similarly, Wei et al. utilized the HOMO energy levels to screen non-sacrificial additives for Zn anode stabilization. Among three anionic surfactants, sodium dodecyl benzene sulfonate (SDBS) possessed the highest HOMO level and strongest electron-donating capability, consistent with its high binding and adsorption energy. This strong coordination with Zn^2+^ helped reduce active water content and enhanced superior adsorption, thereby inhibiting Zn-H_2_O interactions [41]. Notably, LUMO and HOMO levels are key design parameters for cathode material modification. For instance, Ye et al. introduced − CN groups into the organic cathode material Hexaazatrinaphthalene, thereby lowering its LUMO and HOMO energies, increasing the operating voltage, narrowing the energy gap, and enhancing electronic conductivity and charge transport [32].
Nevertheless, it is important to recognize that in practical electrolytes, the electrochemical stability window cannot be solely determined by the HOMO–LUMO gap due to the influence of solvation effects and additives. Although the HOMO and LUMO levels are correlated with redox behavior, an energy offset of several eVs may occur. Therefore, caution should be exercised when using HOMO and LUMO as a direct indicator of the electrochemical stability [42].
Electrolyte Structure Regulation
Zinc Salt Improvements
The selection of zinc salt fundamentally determines the physicochemical properties of the aqueous electrolyte, directly impacting the electrochemical performance of Zn anode in AZIBs. Crucially, the nature of the salt governs electrolyte pH, which strongly influences Zn^2+^ electrodeposition behavior, alongside the parasitic HER kinetics and surface passivation. In detail, the anion size and chemical properties significantly affect the Zn^2^⁺ solvation structure, determining the number of coordinated water molecules, the energy barrier for Zn^2^⁺ desolvation, and the overall ion diffusion dynamics. The intrinsic water solubility of different zinc salts further constrains the maximum achievable salt concentration in the electrolyte. Notably, electrolyte concentration has a profound impact on solvation chemistry: higher concentrations can reconstruct solvation shells, reduce free water activity, increase viscosity, and modulate ion transport. Therefore, rational selection of zinc salt requires a comprehensive balance among key parameters, including pH, anion size, concentration, desolvation kinetics, and ion diffusion capabilities, to achieve reversible Zn plating/stripping and mitigate parasitic reactions.
Conventional zinc salts, such as ZnCl_2_, Zn(ClO_4_)2, Zn(NO_3_)2, Zn(CF_3_SO_3_)2, and Zn(CH_3_COO)2, have been explored, each demonstrating distinct chemical properties and consequently differing impacts on electrolyte pH, solvation structure, and kinetics for the Zn anode (Table 1) [43–49], while these salts still face several challenges, including unstable Zn^2+^ solvation structures, high water reactivity, uneven ion flux, all of which might lead to the parasitic side reactions thereby reducing cycle life and coulombic efficiency. Initially, KOH was employed as the electrolyte due to the faster Zn^2+^ kinetics in alkaline media. However, long-term battery operation is hindered by anode passivation caused by self-corrosion and electrochemical corrosion. In alkaline conditions, the redox potential of ZnO/Zn (− 1.26 V vs. SHE) is more negative than that of the HER (− 0.83 V vs. SHE), accelerating spontaneous zinc oxidation. Continued cycling leads to irreversible zinc loss, forming “dead zinc” and insulating precipitates. The main reactions are summarized below [50]: Table 1. Comparison of the performance of different zinc salts as electrolytesElectrolytesCathodesSolubilityCapacityCycle performanceReferences1 m Zn(NO_3_)2_graphite paper138 g/100 mLLow capacity47.4% CE at 0.02 A g^−1^[64]1 m Zn(CH_3_COO)2_graphite paper30 g/100 mLLow capacity64.8% CE at 0.02 A g^−1^[64]0.5 m Zn(CH_3_COO)2_Na_3_V_2(PO_4)3_30 g/100 mL97 mAh·g^−1^ at 0.5Cretains 74% capacity after 100 cycles[65]Zn(CH_3_COO)2_NH_4_V_4_O_10_30 g/100 mL281mAh g^−1^at 500 mA g^−1^with a capacity retention of 90.13% after 200 cycles[66]3 m Zn(CF_3_SO_3)2_HNaV_6_O_16·4H_2_OHighly soluble444mAh g^−1^at 0.5 A g^−1^with a capacity retention ratio of 93.7% after 1000 cycles (5 A g^−1^)[67]3 m Zn(CF_3_SO_3)2(NH_4)x_V_2_O_5·nH_2_OHighly soluble344 mAh g^−1^at 0.2 A g^−1^with 80% retention after 2,000 cycles at 0.2 A g^−1^[68]3 m Zn(CF_3_SO_3_)2_NH_4-V_2_CT_x_/ZnOHighly soluble173 mAh g^−1^at 2 A g^−1^the average CE is almost 100% during 300 cycles at 2 A g^−1^[69]3 m Zn(CF_3_SO_3_)2_V_2_O_5·2.2H_2_OSoluble in waterhigh initial capacity (4.5 mA h cm^−2^)25,000 cycles at 1 A g^−1^[70]Zn(CF_3_SO_3_)2_CuSoluble in waterHigh capacityCE of 99.9% at 1 mA cm^−2^ and 1 mA h cm^−2^[71]3 m Zn(CF_3_SO_3)2_NH_4_V_4_O_10_Soluble in water/high capacity retention rate of 71.1% after 1000 cycles[72]1 m ZnCl_2_Ca_0.20_V_2_O_5∙0.80H_2_O395 g/100 mL296 mAh g^−1^at 50 mA g^−1^≈50% CE at 1.6 A g^−1^[73]30 m ZnCl_2_Ca_0.20_V_2_O_5_∙0.80H_2_O395 g/100 mL496 mAh g^−1^at 50 mA g^−1^99.6% CE at 1.6 A g^−1^[73]3 m ZnCl_2_NaV_3_O_8_395 g/100 mL362 mAh g^−1^ at 0.2 A g^−1^84 cycles at 1 A g^−1^[74]3 m Zn(ClO_4_)2_NaV_3_O_8_4.30 mol/kg352 mAh g^−1^ at 0.2 A g^−1^30 cycles at 1 A g^−1^[74]1 m Zn(ClO_4)2_VO_2_4.30 mol/kg240 mAh g^−1^at 0.5 A g^−1^high stability for over 3500 h[46]3 m Zn(ClO_4)2_Polyaniline4.30 mol/kg174 mAh g^−1^at 0.5 A g^−1^70,000 cycles at − 35 °C under 15 A g^−1^[75]1 m ZnSO_4_Zn53.8 g/100 mL/98.0% CE at 2 mA cm^−2^[46]3 m ZnSO_4_V_6_O_13 with high-content V^5+^53.8 g/100 mL520 mAh g^−1^at 0.5A g^−1^with 85.3% capacity retention at the 1000th cycle at 2 mA cm^−2^[76]3 m Zn(BF_4_)2_polyaniline and carbon nanotubesHighly soluble109 mAh g^−1^ at 0.1 A g^−1^high capacity retention of 94% after 1000 cycles at 1A cm^−2^[77]3 m Zn(BF_4)_2_NaV_3_O_8_Highly soluble193 mAh g^−1^ at 0.2 A g^−1^56 cycles at 1 A g^−1^[74]
Anode:
\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$${\text{Zn}}^{{{2} + }} + {\text{ 2OH}}^{ - } = {\text{Zn}}\left( {{\text{OH}}} \right)_{{2}} + {\text{ 2e}}^{ - }$$\end{document} \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$${\text{Zn}}\left( {{\text{OH}}} \right)_{{2}} = {\text{ ZnO }} + {\text{ H}}_{{2}} {\text{O}}$$\end{document}Cathode:
\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$${\text{MnOOH }} + {\text{ H}}_{{2}} {\text{O }} + {\text{ e}}^{ - } = {\text{ Mn}}\left( {{\text{OH}}} \right)_{{2}} + {\text{ OH}}^{ - }$$\end{document}Accumulation of byproducts, such as Zn(OH)2, ZnO, and Mn(OH)2, significantly reduces capacity and coulombic efficiency, resembling a “primary battery” [51]. Moreover, in high-concentration 6 M KOH electrolyte, the battery experienced rapid polarization within six cycles and short-circuited in 5.3 h due to aggressive dendrite formation [52]. These findings highlight the importance of electrolyte pH on battery stability.
In neutral and mildly acidic electrolytes, strong Coulombic interactions between the solvated Zn^2+^ ion and its surrounding H_2_O shell accelerate parasitic water reduction; meanwhile, the HER kinetics become highly pH-dependent. HER dominates in acidic media (2H^+^ + 2e⁻ → H_2_), competing with Zn^2+^ deposition. H_3_O⁺ ions first absorb and accept electrons on the anode surface (Volmer step), forming H* intermediates. These either recombine to form H_2_ (Tafel step) or react with more H_3_O⁺ and electrons (Heyrovsky step). In neutral conditions, the HER mechanism is more complex and influenced by multiple factors, including electrolyte concentration and species identity [53]. Zinc possesses a relatively high hydrogen evolution overpotential, which theoretically suppresses HER. This hydrogen evolution overpotential (η) can be expressed using the Tafel Equation:
\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$\eta =a+b\text{log}i$$\end{document}where i is the current density, and a and b are constants ( b is the Tafel slope). The Tafel slope is often used to identify the rate-determining step of the HER. Despite a high value of a, HER still occurs spontaneously due to practical kinetic factors, including electrode roughness, temperature, and electrolyte composition [54]. During charging, HER continuously consumes both electrons and electrolyte, reducing coulombic efficiency and shortening battery life. Accumulated H_2_ also elevates internal pressure, thereby causing battery swelling and potentially even explosion [55, 56].
To mitigate these issues, near-neutral zinc salts are preferred for stable cycling. Zn(CF_3_SO_3_)2 has gained popularity due to its effective desolvation capability (Fig. 3a) [49]. Nevertheless, other near-neutral zinc salts like Zn(NO_3_)2 and Zn(ClO_4_)2 also pose challenges. Their strong oxidizing anions can induce ZnO passivation on the anode, raising Zn^2+^ dissolution/deposition impedance and slowing kinetics. Interestingly, in a Zn(ClO_4_)2 and NaClO_4_ mixed system, high ClO_4_^–^ concentrations replace water in the solvation shell, reducing free water molecules and thus suppressing HER, while also modifying ion transport properties (Fig. 3b–d) [48]. Though Cl^−^ species are less oxidizing and can coordinate with Zn^2+^ to form ZnCl_4_^2−^ complexes (Fig. 3e, f) [47], AZIBs with ZnCl_2_ still suffer from poor cycling due to a limited electrochemical stability window [43]. In contrast, Zn(CF_3_SO_3_)2, and Zn(TFSI)2 are considered promising candidates owing to their wide electrochemical windows, high compatibility with electrode materials, and their anions that promote beneficial physicochemical properties like reduced solvation and suppressed water activity [57, 58].Fig. 3a Three-dimensional molecular dynamics snapshots of the Zn(CH_3_SO_3_)2 system, with partially enlarged view illustrating the solvation structure of Zn^2+^ [49]. Equilibrium trajectories snapshots from ab initio MD simulations of b 0.5 mol kg^−1^ Zn(ClO_4_)2, c 0.5 mol kg^−1^ Zn(ClO_4_)2 + 9 mol kg^−1^ NaClO_4_, and d 0.5 mol kg^−1^ Zn(ClO_4_)2 + 18 mol kg^−1^ NaClO_4_ [48]. e Representative solvation structure of Zn^2+^ ions. f Schematic showing how the introduction of Cl^−^ ions releases coordinated water molecules, resulting in a modified solvation structure [47]. g A schematic illustration of the growth of Zn dendrites in aqueous electrolytes. h Proposed formation mechanism of ZnF_2_-Zn_5_(CO_3_)2(OH)6-organic SEI [59]. XPS of i N 1* s*, j C 1* s*, and k F 1* s* with different Ar sputtering durations [59]. l CV of Zn||MnO_2_ full cells at a scan rate of 0.1 mV s^−1^ [59]. m The radial distribution function and coordination number plots of Zn^2+^solvation structure in ZnSO_4_ and Zn(TFSI)2 electrolyte [63]
For instance, Li et al. introduced trace amounts of NO_3_^−^ into the Zn(CF_3_SO_3_)2 electrolyte, which facilitates salt decomposition and promotes the formation of a compact SEI (Fig. 3g, h). Ar sputtering-assisted X-ray photoelectron spectroscopy (XPS) analysis revealed that the SEI comprises an organic-rich outer layer and an inorganic ZnF_2_-rich inner layer (Fig. 3i–k). Additionally, cyclic voltammetry measurements demonstrated the effect of the SEI on reducing polarization voltage(Fig. 3l) [59]. In Zn(CF_3_SO_3_)2, the bulky CF_3_SO_3_^−^ anions effectively reduce Zn^2+^ solvation by decreasing the number of coordinated water molecules, thereby facilitating Zn^2+^ transport and improving the coulombic efficiency [60, 61]. Furthermore, CF_3_SO_3_^−^ anions can react with Zn anode to form a stable SEI, contributing to long-term cycling stability [62]. Similarly, Zn(TFSI)2, a commonly studied zinc salt, features a large anionic structure that disrupts strong hydrogen bonding and reduces the population of free water molecules in the electrolyte (Fig. 3m). While TFSI^−^ ions effectively suppress aqueous side reactions and promote Zn^2+^ transport, their widespread application remains limited by the high cost of the salt [63]. Beyond the type of zinc salt, increasing the electrolyte concentration is also considered an effective strategy to mitigate side reactions by regulating the interaction between Zn^2+^ and H_2_O. Table 2 summarizes the ionic conductivity, electrochemical stability window, and cycling performance of electrolytes across various concentrations, underscoring the significance of concentration engineering. Recent studies demonstrate that elevated salt concentrations enhance electrostatic interactions between anions and cations while weakening the coordination between Zn^2+^ and H_2_O. This adjustment reshapes the solvation environment and solvent sheath surrounding Zn^2+^ in the electrolyte [78, 79]. As a result, the hydration coordination number of Zn^2+^ is reduced to below 6, thereby suppressing the HER [79, 80]. For instance, in the ZnCl_2_-LiCl system, increasing LiCl concentration reduces Zn^2+^ hydration. Molecular dynamics simulations reveal that a higher Li^+^ concentration increases the number of water molecules coordinated to Li^+^, consequently decreasing the population of free water molecules and lowering HER probability (Fig. 4a–c) [78]. Similarly, in the Zn(TFSI)2-LiTFSI electrolyte, increasing LiTFSI concentration alters the Zn^2+^solvation structure, with the solvation sheath being predominantly occupied by TFSI^−^ anions in highly concentrated formulations (Fig. 4d) [52]. Huang et al. compared two electrolyte systems with different concentrations: a high-concentration system (4.2 m ZnSO_4_·7H_2_O + 0.1 m MnSO_4_·H_2_O, denoted as CZSAE) and a low-concentration system (2 m ZnSO_4_·7H_2_O + 0.1 m MnSO_4_·H_2_O, denoted as LZSAE). O 1* s* XPS spectra revealed that a passivation layer enriched in S–O species dominates in CZSAE, indicating increased Zn^2+^ coordination with sulfate anions and reduced interaction with water molecules(Fig. 4e, f). This solvation adjustment directly influences the composition and morphology of the passivation layer (Fig. 4g, h) [81]. Table 2. The effects of different electrolyte concentrations on various battery performanceElectrolytesESW (V)Ionic ConductivityDeviceCycling PerformanceReferences5 m ZnCl_2_1.6HighZn||ZnCE of 73.2%[82]10 m ZnCl_2_2.0HighZn||Zn/[82]15 m ZnCl_2_1.612 mS cm^−1^Zn||K_0.486_V_2_O_5_1400 cycles with 95.02% capacity retention[85]15 m ZnCl_2_ + 1 m LiCl2.4/Zn||LiFePO_4_a capacity retention of 70% after 1000 cycles at 3C[86]20 m ZnCl_2_2.2LowZn||Zn/[82]30 m ZnCl_2_2.33 mS cm^−1^Zn||ZnCE of 95.4%[82]21 m LiTFSI + 2 m Zn(OTf)2− 0.102 /2.745 of HER/OER0.24 of tZn^2+^ valueZn||YP-80Fcycle 5220 hat 0.5 A g^−1^[87]21 m LiTFSI + 3 m Zn(OTf)2_2.67 mS cm^−1^Zn||GraphiteCE of 95% over 600 cycles at 0.2 A g^−1^[88]1 m Zn(OTf)2 + 20 m LiTFSI2.91.71 mS cm^−1^Zn||LiMn_2_O_4_CE of 99.62% at 300 cycles[89]30 m KAc + 3 m LiAc + 3 mZnAc_2_2.36.5 mS cm^−1^Zn||LiFePO_4_1000 h at 0.1 mA cm^–2^[90]1.5 m LiAc + 1.5 m ZnAc_2/HighZn||LiMn_2_O_4_cannot provide sufficient cyclability[90]30 m KAc + 1 m ZnAc_2_2.2HighZn||AChigh capacity retentionover 10 000 cycles[91]1 m KAc + 1 m ZnAc_2_1.6LowZn||ACrapid capacity fading occurs within 10 cycles[91]1 m Zn(OTf)2_2.3HighZn||ZMO/Calong with a lower CE after the 3rd cycle[43]3 m Zn(OTf)2_2.53.47 S cm^–1^Zn||ZMO/Cnearly 100% CE for both anode and cathode[43]0.5mZn(ClO_4)2 + 18 m NaClO_4/98.5 mS cm^−1^Zn||NVOCapacity retention of 80.9% after 15 days at 0.1 A g^−1^[48]8 m Zn(ClO_4_)2_2.8/Zn||Graphitecycle life of over 500 cycles at 0.1 A cm^–2^[92]3 m Zn(ClO_4)2_2.54.23 mS cm^−1^Zn||MnO_2_cycle life of over 1000 cycles at 6 A g^−1^[93]Fig. 4. Molecular dynamics snapshots of electrolytes at various salt concentrations: a 30 mol kg^–1^ ZnCl_2, b 30 mol kg^–1^ ZnCl_2_ + 5 mol kg^–1^ LiCl, c 30 mol kg^–1^ ZnCl_2_ + 10 mol kg^–1^ LiCl [78]. d Zn^2+^ solvation structures in electrolytes containing 1 mol kg^–1^ Zn(TFSI)2 with varying concentrations of LiTFSI (5, 10, and 20 mol kg^–1^) [52]. XPS investigation of O 1 s spectra showing the passivation layer formation on Cu current collectors after Zn deposition at the fifth cycle using e LZSAE and f CZSAE electrolytes [81]. Schematic illustrations of the passivation layer formation and solvation structure during Zn plating on the Cu substrate using g LZSAE and h CZSAE [81]. i Schematic diagram of the electrochemical stability window [79]. j CV curves of 7.5 m ZnCl_2_ + PAM HEs solution under two different voltage windows [83]
Additionally, recent studies have shown that a high concentration of 30 m ZnCl_2_ effectively suppresses the formation of fully hydrated [Zn(H_2_O)6]^2+^ complexes and instead promotes the formation of ion pairs, such as [Zn(H_2_O)Cl_4_]^2−^ and [ZnCl_4_]^2−^, more than in 20 m ZnCl_2_. Experimental results indicate that the electrochemical stability window significantly broadens with increasing ZnCl_2_ concentration, suggesting suppression of HER and the formation of inactive by-products, such as Zn(OH)2 and ZnO (Fig. 4i) [82]. To address the dissolution of Zn-based ion clusters in aqueous ZnCl_2_ solutions, Wang et al. developed a water-in-salt (WIS) hydrogel electrolyte containing 7.5 m ZnCl_2_. This system not only broadened the electrochemical window but also enhanced the reversibility of the Zn metal anode (Fig. 4j) [83]. Notably, the battery retained 95.1% of its initial capacity after 100,000 charge–discharge cycles. However, elevated electrolyte concentration can introduce several drawbacks, including reduced ionic conductivity, increased viscosity, and high voltage polarization, all of which hinder battery performance in practical applications [84]. Therefore, careful optimization of the electrolyte concentrations for each zinc salt is essential to balance performance, stability, and practical applicability in AZIBs.
Electrolyte Additives
Electrolyte additives fundamentally modify the chemical and physical environment at the anode/electrolyte interface in a “trace amounts yet highly effective” manner, playing a crucial role in interfacial stability in AZIBs [94]. To function effectively, ideal electrolyte additives must possess high water solubility, a critical prerequisite that guides the screening and rational design of additive molecules. The hydrophilic and hydrophobic balance of functional groups, along with potential non-covalent interactions, should be comprehensively considered to ensure both adequate dissolution and targeted interaction at the anode/electrolyte interface. Moreover, strategies, such as pH adjustment and the use of solubilizing agents, are widely employed to enhance additive water solubility [95, 96]. Electrolyte additives must also demonstrate chemical compatibility with other battery components and avoid inducing harmful side reactions. From a practical standpoint, additives should be low-cost and exhibit minimal toxicity to meet scalability and safety requirements [97].
Based on these design principles and their mechanisms of action, commonly employed electrolyte additives for AZIBs can be broadly categorized into five groups: (i) self-healing via electrostatic shielding, (ii) surface adsorption, (iii) desolvation regulation, (iv) in situ SEI formation layer, and (v) crystal plane regulation. A detailed discussion of these mechanisms and representative examples will be presented below.
Self-Healing Electrostatic Shield Mechanism
The self-healing electrostatic shielding mechanism was initially proposed to enhance the performance of lithium-ion batteries. Zhang and co-workers observed that alkali-metal ions, such as Cs⁺ and Rb⁺, preferentially adsorb onto lithium-dendrite tips, forming a positively charged protective layer that redirects Li⁺ deposition toward the surrounding surface. This electrostatic shield repels incoming Li⁺ from the protrusion tip, forcing uniform deposition in adjacent regions and thereby “self-healing” nascent defects, thereby suppressing dendrite growth; this process is termed the self-healing electrostatic shield mechanism [98]. Theoretical calculations further indicate that these ions possess lower adsorption energies and diffusion barriers on lithium metal, facilitating their migration and selective accumulation at dendritic tips. In addition, they also show a tendency to prioritize the repair of SEI defects in battery systems [99].
Inspired by these findings, the strategy has been extended to AZIBs, yielding promising results. Monovalent and multivalent cations, such as Li^+^ [100], Na^+^ [101, 102], Mg^2+^ [103], Ga^3+^ [104], Rb^+^ [105], and plasma-derived additives, have been reported to form positively charged electrostatic shields around zinc deposition tips, effectively suppressing dendritic growth. For example, Yuan et al. achieved a high coulombic efficiency in Zn||Zn symmetric cells by introducing lithium halides into the electrolyte [100]. DFT calculations indicated that the adsorption energy of Zn^2+^ ion the surface of Zn (100) surface is lower than that on the Zn (200) surface [106], causing Zn^2+^ to preferentially adsorb onto the (100) face and form daisy-chain dendrites (Fig. 5c). Experimental results demonstrated that Li^+^ ions were found to preferentially adsorbed onto the (100) face due to their low binding energy, redirecting zinc nucleation toward the (002) plane and resulting in a denser, smoother anode surface (Fig. 5d).Fig. 5a Schematics illustrations of Zn deposition process in 2 M ZnSO_4_(top row) and ZnMg-0.1 (bottom row) electrolyte [103]. b Diagram depicting the effect of Ga^3+^ ion additives on Zn deposition process [104]. c Illustration of the tip effect, wherein Zn^2+^ ions preferentially adsorb to the tips and edges of the existing Zn deposit [100]. d Schematic showing that lithium ions preferentially adsorb on the Zn (100) plane, guiding Zn^2+^ deposition toward the (002) plane and suppressing dendritic growth [100]. In situ optical microscopic images of Zn^2+^ deposition on Zn foils at a current density of 20 mA cm^−2^ in e ZnSO_4_ (2 M) and f ZnSO_4_ (2 M) + 1.5 mM Rb_2_SO_4_ electrolytes [105]
Distinct from the Li^+^, Na^+^, and K^+^ ions, Rb^+^, with a larger ionic radius and stronger electrostatic repulsion, more effectively occupies surface protrusions, forming a broader electrostatic shield and promoting lateral Zn^2+^ deposition across the Zn surface [105]. DFT results show that Rb^+^ exhibits a significantly higher adsorption energy on metallic Zn (− 32.60 kcal mol^−1^) compared to H_2_O molecules (− 3.55 kcal mol^−1^), confirming its preferential adsorption. In addition, electrolytes containing Rb^+^ demonstrated a lower EDL capacitance(159.45 μF cm^−2^) relative to pristine 2 M ZnSO_4_ (216.09 μF cm^−2^), attributed to Rb_2_SO_4_ occupying active tip sites and altering Zn^2+^adsorption behavior (Fig. 5e, f). Thus, the Zn||Zn symmetric cells with 1.5 mmol Rb_2_SO_4_ exhibited a prolonged cycle life exceeding 6000 h at a current density of 0.5 mA cm⁻^2^ and a capacity density of 0.25 mAh cm^−2^, superior to the control group without Rb_2_SO_4_ (which cycled for 300 h).
Additionally, Wu et al. introduced a cost-effective gelatin molecule as an additive in ZnSO_4_ electrolyte [49], in which the self-healing electrostatic shield mechanism was also achieved. Owing to their steric hindrance, gelatin molecules preferentially adsorbed on the Zn surface during the electrochemical cycling. The strong Zn^2+^ binding capacity of gelatine, combined with positively charged − CN_3_H_5_^+^ groups generated under mildly acidic conditions, the 2D Zn^2+^ diffusion was effectively inhibited. This facilitates uniform zinc deposition and enhanced interfacial stability, thereby improving overall battery performance.
Adsorption Effects
In AZIBs, the adsorption effect of additives is considered an effective strategy to reduce the two-dimensional diffusion time of Zn^2+^ ions and lower the nucleation overpotential, thereby inhibiting the dendrite growth and mitigating side reactions. Additives with strong adsorption capabilities can form a uniformly distributed protective layer on the Zn metal surface, preventing direct contact between the anode and electrolyte. This physical barrier significantly suppresses parasitic HER and promotes uniform Zn deposition during electroplating [107]. In contrast to the self-healing electrostatic shield mechanism, which involves a repulsive layer created by cations with the same charge as Zn^2+^, neutral or even negatively charged species can also induce an adsorption effect. These species increase the density of nucleation sites and reduce the energy barrier for Zn^2+^ nucleation, thereby promoting homogeneous metal deposition. Notably, certain additives can guide Zn^2+^ deposition along a preferred crystal orientation, optimizing the microstructure of the zinc layer and enhancing electrochemical performance.
Among the reported additives, graphene-based materials have emerged as a particularly promising material. Qiu et al. [108] incorporated graphene oxide (GO) into the electrolyte and demonstrated that GO particles strongly adsorb onto the Zn surface through electrostatic interactions. This adsorption suppresses local electric field inhomogeneities and facilitates a more uniform field distribution, thereby lowering the Zn^2+^ nucleation overpotential. Furthermore, the abundant polar functional groups on GO surfaces accelerate Zn^2+^ migration to the reaction interface, increasing active nucleation sites and enhancing ion diffusion, resulting in a denser and smoother zinc deposit. As a result, the AZIBs using GO additive exhibited remarkable stability, operating continuously for over 650 h at a current density of 1 mA cm^−2^ (Fig. 6a), and maintaining stable cycling for 400 and 140 h at 5 mA cm^−2^ (Fig. 6b) and 10 mA cm^−2^, respectively (Fig. 6c). Electrochemical impedance spectroscopy (EIS) analysis revealed significantly reduced resistance in GO-containing systems (Fig. 6d), indicating improved charge transfer kinetics. Additionally, the system achieved a high coulombic efficiency of 99.16%, which is superior to the conventional ZnSO_4_ electrolyte in both cycling stability and energy conversion efficiency.Fig. 6. Electrochemical investigation of Zn anodes employing GO electrolyte additive [108] at a 1 mA cm^−2^ of 0.5 mAh cm^−2^, b 5 mA cm^−2^ of 2.5 mAh cm^−2^, c 10 mA cm^−2^ of 5 mAh cm^−2^. d EIS analysis of the Zn anodes with and without GO additives. e Schematic illustration of surfactant-modulated interactions between Zn anodes and Zn^2+^ ions, accompanied by the corresponding plating morphologies [109]. f Long-term cycling stability of Zn||Zn symmetric cells with a depth of 2 mAh cm^−2^ at 4 and 10 mA cm^−2^ in various surfactant-modulated electrolytes [109]. g Cycling performance of Zn||MnO_2_ batteries in various surfactant-modulated electrolytes at a current density of 1 A g^−1^ [109]. h Raman spectra of the electrolyte with 6-AA in the Raman shift range of 965–1005 cm^−1^ [113]. i pH values of the pristine electrolyte and the various concentrations of the 6-AA added electrolyte [113]
Building upon insights into the rate of change of electrochemical surface area (dS/dt), Xie et al. developed two nucleation models, the instantaneous nucleation model (INM) and the continuous nucleation model (CNM). The INM describes limited two-dimensional diffusion under conditions of weak electrostatic adsorption of Zn^2+^, while CNM occurs when Zn^2+^ adsorption is enhanced, facilitating continuous two-dimensional diffusion and nucleation (Fig. 6e) [109]. The authors demonstrated that ZnSO_4_ electrolytes, due to the relatively weak electrostatic adsorption with Zn^2+^, favor the INM. In this scenario, multiple independent nuclei form rapidly at the onset of electrodeposition and subsequently grow via three-dimensional diffusion. However, this leads to a loosely packed zinc layer prone to dendrite formation, as evidenced by prominent dendrite growth when cetyltrimethylammonium bromide (CTAB) was used as an additive. In contrast, the introduction of sodium dodecylbenzene sulfonate (SDBS) into the electrolyte led to continuous nucleation, with new crystal nuclei forming both during the early and later stages of deposition (Fig. 6f, g). This behavior indicates enhanced Zn^2+^ electro-adsorption and supports a CNM mechanism, which promotes denser and more planar Zn deposition through a dominant two-dimensional diffusion pathway. This continuous nucleation mechanism has also proven effective for other long-chain anionic surfactants, such as the anionic part of sodium dodecyl sulfate (SDS) [110], and provides a promising strategy for the rational design of Zn anodes [111].
Remarkable adsorption properties were also observed in electrolytes containing ethylenediaminetetraacetic acid (EDTA) anions. DFT calculations indicated the adsorption energy of EDTA anions on the Zn surface is − 1.62 eV, significantly lower than that of free H_2_O (− 0.31 eV) and the Zn(H_2_O)6^2+^ complex (− 0.12 eV), indicating a stronger affinity of the Zn surface for EDTA anions. Owing to their firm adsorption, EDTA anions effectively block HER sites on Zn surface, thereby suppressing parasitic HER during both plating and stripping processes. Furthermore, the strong complexation between EDTA and Zn^2+^ provides additional nucleation sites during the initial deposition stage, which effectively facilitates grain refinement. The adsorption layer formed by EDTA also facilitates the formation of nanoscale ion-transport pathways, preventing direct water access and mitigating H_2_O interference in the deposition process. These combined effects contribute to uniform and flat zinc deposition and enhance the cycling stability. Experimental results indicated that a symmetric cell with tetrasodium EDTA achieves over 2000 h of stable cycling at current densities of 5 mA cm^−2^, approximately 30 times longer than the pristine ZnSO_4_ electrolyte [111]. Besides, Wang et al. demonstrated that 0.04 M EDTA, when added to a zinc-vanadium pentoxide battery system, improved the coulombic efficiency to 99.36% and enabled over 4000 h of stable operation, confirming EDTA’s effectiveness in boosting AZIBs’ performance [112]..
Similarly, the zwitterionic molecule 6-aminocaproic acid (6-AA) has shown potential to enhance battery performance through surface adsorption effects [113]. As illustrated by the Raman shift in Fig. 6i, the cationic portion (amino group) of 6-AA can interact with Zn^2+^ ions on the Zn metal surface via physical adsorption or chemical bonding, creating a barrier that inhibits water molecules and other potential reactive ions’ access and reduces side reactions.
Desolvation
During the electrodeposition of Zn^2+^, the desolvation of hydrated zinc ions of [Zn(H_2_O)6]^2+^ is a critical step that significantly affects Zn deposition behavior. To enable long-term stable performance of AZIBs, this process, requiring partial or complete removal of water molecules from the solvation shell, must effectively reverse the initial solvation of Zn^2+^ [114, 115]. Notably, the water molecules released during the desolvation process are often more reactive than free water [116], which increases the energy barrier for desolvation, and subsequently hinders Zn^2+^ diffusion and deposition kinetics [117]. In addition to slowing ion transport, these highly reactive water molecules could attack the cathode lattice structure, leading to cathode dissolution, irreversible phase transitions, and overall degradation of electrochemical stability. Thus, regulating the desolvation behavior of [Zn(H_2_O)6]^2+^ by rational electrolyte design is essential to achieve uniform and efficient zinc deposition [118, 119].
Recent advances in electrolyte engineering have made significant progress by tailoring the Zn^2+^ solvation structure. Ma et al. focused on manipulating the outer solvation shell through 2-propanol additives, which establish a eutectic solvent network that preferentially interacts with Zn (101) crystal planes (Fig. 7a) [120]. The 2-propanol-water eutectic structure exhibits strong directional affinity toward the Zn (101), promoting oriented Zn deposition and accelerating desolvation kinetics, particularly at low temperature conditions, thus delivering fast and reliable electrochemical performance. Moreover, 2-propanol disrupts the pre-existing H_2_O − H_2_O hydrogen bonding network by forming new 2-propanol − H_2_O hydrogen bonds. Its hydroxyl groups also serve as coordination sites for Zn^2+^, contributing to the formation of efficient ion transport pathways and enhancing Zn^2+^ mobility.Fig. 7a Schematic showing the participation of 2-propanol in the second solvation shell of Zn^2+^ ions [120]. b Electrostatic potential mapping of the HA molecule. c Raman spectra for pristine ZnSO_4_ and ZnSO_4_ − HA with varying amounts of HA. Long-term cycling stability comparison for d pristine ZnSO_4_ and e ZnSO_4_ − HA electrolyte systems [121]. f Schematic illustration of the solvated Zn^2+^ deposition behavior in pristine Zn(OTf)2 and PSCA/Zn(OTf)2 hydrogel electrolyte [122]. g Cycling tests of the Zn||Zn cells in various electrolytes at 1 mA cm^−2^ and 0.5 mAh cm^−2^ [122]
Qiu et al. introduced an innovative strategy by incorporating hyaluronic acid (HA), a chaotropic polymer-type additive, into the electrolyte, which effectively suppressed undesired dendrite growth and side product formation on the Zn anode (Fig. 7b) [121]. As shown in Raman spectra (Fig. 7c), the HA additive disrupts the original hydrogen-bonded network of H_2_O−H_2_O interactions, reducing the number of active water molecules. This disruption leads to the formation of new HA−H_2_O hydrogen bonds, thereby mitigating parasitic reactions driven by highly reactive water species. Additionally, abundant functional groups, such as carboxyl (−COOH) and hydroxyl (−OH), distributed along the HA polymer chains serve as binding sites for Zn^2+^, facilitating the formation of effective ion transport channels and enhancing Zn^2+^ transfer. The dynamic coupling/decoupling behavior of Zn^2+^ along the flexible HA chains elevates the Zn^2+^ transference number to 0.62, significantly improving the cycling life of Zn||Zn symmetric cells. As a result, the constructed AZIBs achieved outstanding cycling stability, with lifetimes of 2,200 h at 1 mA cm^−2^/1 mAh cm^−2^ (Fig. 7d) and 800 h at 5 mA cm^−2^/5 mAh cm^−2^ (Fig. 7e), respectively.
To further regulate the hydration structure of Zn^2+^, a zincophilic anionic hydrogel electrolyte (PSCA/Zn(OTf)2) was developed, incorporating dodecyl sulfate anions ((OSO_3_R)⁻) micelles to suppress dendrite growth and parasitic reactions on the Zn anode (Fig. 7f) [122]. Experimental evidence revealed that (OSO_3_R)⁻ anions disrupt the native H_2_O − H_2_O hydrogen-bond network, reducing the population of active water molecules, and form new (OSO_3_R)⁻-H_2_O hydrogen bonds. This suppresses passivation and HER. Furthermore, functional groups along the (OSO_3_R)⁻ chains provide Zn^2+^ binding sites, enabling the construction of effective ion transport pathways and enhancing Zn^2+^ transference. This design significantly improves the lifetime of Zn||Zn symmetric cells, achieving 760 h at 1 mA cm^−2^/0.5 mAh cm^−2^. Additionally (Fig. 7g), dynamic ion-polymer interactions along flexible (OSO_3_R)⁻ chains further promote Zn^2+^ mobility, resulting in ultra-long cycling stability of over 40,000 cycles with a capacity decay of only 0.00027% per cycle.
In addition to the small-molecule additives, polymeric additives also possess unique capabilities in modulating Zn^2+^ solvation structure [95]. He et al. employed a nonionic amphiphilic polymer additive (APA) to establish a nano-scaled hydrophobic confinement layer at the anode–electrolyte interface. The hydrophilic acrylamide segments of APA preferentially adsorb onto the Zn metal surface, while the hydrophobic methacrylate segments form a water-repellent shell that locally suppresses H_2_O activity. This amphiphilic structure induces a partial desolvation of Zn^2+^ by replacing coordinated water molecules with polymer segments, thereby minimizing water-related side reactions. This strategy achieves exceptional cycling stability, exceeding 8,800 h at 1 mA cm^−2^/1 mAh cm^−2^ [123]. Similarly, Zhang et al. introduced poly(acrylic acid) (PAA) to reconstruct the Zn^2+^ solvation structure from [Zn(H_2_O)6]^2+^ to a more conductive [Zn(H_2_O)2(AA)4]^2−^ complex, thereby accelerating charge-transfer kinetics. The carboxyl moieties in PAA self-assemble into a hydrophobic interfacial layer that isolates the Zn anode from bulk water, promoting Zn^2+^ desolvation and uniform electrodeposition, ultimately enhancing cycling reversibility [124].
Formation of In Situ SEI Layer
The formation of the SEI is closely associated with the decomposition of solvents and additives in the electrolyte. Structurally, the SEI typically comprises two distinct layers: a dense inner layer and a porous outer (diffuse) layer. The dense layer is primarily composed of elements, such as Zn, F, and O, possessing high mechanical strength and compactness; whereas the outer layer mainly consists of organic compounds, offering flexibility and porosity. This dual-layer architecture effectively mitigates direct contact between the Zn anode and the electrolyte, thereby mitigating side reactions and dendrite growth. Furthermore, the intimate adhesion between the diffuse and dense layers prevents SEI cracking and facilitates Zn^2+^ ions transport. However, in aqueous electrolytes, the formation of an in situ SEI layer remains challenging due to intense HER and the difficulty in decomposing zinc salts [125]. Current efforts focus on constructing artificial SEI layers using inorganic or organic composite coatings (e.g., MOFs and COFs) or introducing trace electrolyte additives [9, 122, 126–129]. Among these approaches, electrolyte additives are particularly attractive due to their operational simplicity, low cost, and compatibility with commercial-scale applications.
Inspired by the phosphating solution Zn(H_2_PO_4_)2 in the electroplating, where a phosphide layer forms on Zn, Guo et al. introduced the Zn(H_2_PO_4_)2 into a ZnSO_4_ electrolyte to induce the in situ formation of an inorganic Zn_3_(PO_4_)2 SEI layer, significantly enhancing the reversibility of the Zn anode [130]. This dense, inorganic-rich SEI acts as an effective physical barrier, preventing water penetration and thus suppressing the HER and passivation. The OH⁻-mediated formation of the SEI endows it with strong Zn^2+^ adsorption capacity, substantially higher than that of the bare zinc anode (Fig. 8a), thereby increasing the Zn^2+^ transference number (Fig. 8b) and reducing the diffusion energy barrier (Fig. 8c). Beyond phosphate-based layers, fluorinated SEI has also been extensively studied for AZIBs. For instance, Cao et al. employed Me_3_EtNOTf as an additive in the Zn(OTf)2 electrolytes, leading to the in situ formation of a 64 nm ZnF_2_ layer, as confirmed by transmission electron microscopy. This layer acts as an electronic barrier to suppress water reduction, while permitting Zn^2+^ transport [131]. Luo et al. constructed a multifunctional in situ SEI composed of PNM-ZnF_2_-ZnS-ZnSO_x_ that blocks water and anion penetration, while enabling selective Zn^2+^ shuttling [132]. Other strategies have also proven effective [133–136]. Wu et al. constructed a dense SEI layer through chelation-induced polymerization of Zn^2+^ with poly(N-[2-(3,4-dihydroxyphenyl)ethyl]-2-methylacrylamide) [137]. Huang et al. introduced saccharin (Sac) as a zincophilic additive. The Sac anion strongly adsorbs onto the Zn surface, forming an electric double layer that repels H_2_O and regulates Zn^2+^ diffusion, thereby suppressing dendrite growth and limiting the water access essential for HER and subsequent passivation [138].Fig. 8a Calculated interaction models of Zn^2+^ ion with the Zn_3_(PO_4_)2·4H_2_O SEI layer (left) and bare zinc anode surfaces (right), respectively [130]. b Comparison of Zn^2+^ ion transference numbers between bare Zn anode and Zn_3_(PO_4_)2 4H_2_O SEI-coated Zn anode [130]. c Calculation of the corresponding migration energy barriers in the Zn_3_(PO_4_)2 4H_2_O SEI layer [130]. d Schematic of real-time dynamic interphase reconstruction with C_3_N_4_QDs as the additives. e Failure mechanism of stationary interface protective coatings [15]. f Schematic diagram of Zn plating and corrosion in pristine, GQD-based and Cl-GQD-based ZnSO_4_ electrolytes [139]. g Schematic diagram of the morphological evolution of the Zn anode in Cl-GOD-based ZnSO_4_ electrolyte [139]. h Binding energies of a water molecule to the Zn (002), (100), (101) crystal plane, and different functional groups in GQDs [139]
However, the in situ formation of the SEI is inherently uncontrollable, making it difficult to precisely regulate its structure, composition, thickness, and compactness. Moreover, current strategies for constructing interfacial layers often suffer from limitations during battery cycling, such as layer fracture, inability to regenerate, or the continuous consumption of electrolyte and functional components. Given that the deposition/stripping of Zn^2+^ ions at the reaction interface is a dynamic process, relying on static interfacial modifications is insufficient to accommodate long-term changes in interfacial microstructure and local electrochemical microenvironments. Therefore, establishing a dynamically stable and compact interface that enables real-time, directional control of interfacial reactions is essential. To address this, our team employed graphitic carbon nitride quantum dots (C_3_N_4_QDs) as an example to evidence this concept of constructing a dynamic self-repairing protective interface [15]. These C_3_N_4_QDs act as highly efficient colloidal ion carriers in the electrolyte, exhibiting strong interactions with Zn^2+^, thereby optimizing their solvation structure and suppressing water activity. Additionally, C_3_N_4_QDs self-assemble horizontally on the Zn anode to form a protective SEI layer. Their periodic sub-nanometer pores are oriented perpendicular to the electrode surface, forming directional ion channels that selectively allow the transport of Zn^2+^ ions while excluding solvated species, effectively achieving a molecular sieving effect. Upon electric field reversal, the C_3_N_4_QDs that were previously tightly attached to the surface of the Zn anode can desorb and return to the bulk electrolyte via coulombic interactions (Fig. 8d, e). This dynamic regeneration occurs repeatedly during each charge–discharge cycle. Consequently, defects or early stage dendrite formation can be repaired in subsequent cycles through C_3_N_4_QDs redistribution. This mechanism actively corrects Zn deposition behavior, fundamentally eliminating the irreversible interface rupture and maintaining long-term interfacial stability. Similarly, Wang et al. introduced positively charged chlorinated graphene quantum dots (Cl-GQDs) as additives. During charging, Cl-GQDs electrostatically adsorb onto the Zn surface, forming a shielding layer. Their chlorine groups exhibit hydrophobic characteristics that further promote uniform Zn deposition (Fig. 8f, g). Upon discharge, Cl-GQDs undergo dynamic regeneration and redissolve into the electrolyte, ensuring a continuous and stable interfacial layer [139]. Theoretical calculations and electrochemical characterizations revealed that Cl-functionalized GQDs exhibit significantly lower binding energies with water molecules (Fig. 8h), confirming their superior hydrophobicity and suitability for constructing robust interfacial layers. Notably, this dynamic self-healing process does not consume the additives, enabling sustained interfacial protection and contributing to significantly enhanced performance of AZIBs.
Regulation of the Zinc Crystal Plane
Understanding the crystallographic behavior of the Zn anode, particularly the distinct electrochemical properties of its crystal facets, is essential for designing and evaluating interfacial strategies in electrolyte engineering. Electrolyte components can markedly influence Zn nucleation and growth orientation via preferential adsorption, thereby dictating deposition morphology and long-term stability [140, 141]. A central goal in optimizing Zn-deposition morphology is to promote preferential growth along the (002) crystal plane. This facet orientation plays a critical role in determining corrosion resistance and the dendrite growth direction of the zinc anode. Crystallographically, during the initial nucleation stage, Zn atoms tend to nucleate on low-energy planes such as Zn (101) and Zn (100) due to the polycrystalline nature of zinc and the anisotropy inherent in its hexagonal close-packed (hcp) lattice. As deposition proceeds, Zn atoms commonly adopt a two-dimensional hexagonal morphology with sharp edges and continue to grow along these crystal planes [142]. In general, a larger angle between the growth direction of Zn dendrites and the anode surface correlates with increased dendrite formation. Under constant voltage, the current response for Zn (002) remains stable after an initial brief two-dimensional diffusion phase, indicative of a steady three-dimensional diffusion process (Fig. 9a). This behavior suggests that the Zn (002) crystal plane exhibits a lower nucleation overpotential and reduced energy barrier for critical nucleus formation (Fig. 9b), making it an ideal target for electrolyte additives aiming to induce uniform nucleation and suppress dendrite formation. The planar symmetric structure and close-packed nature of the (002) crystal plane facilitate the uniform and dense deposition of zinc atoms, effectively eliminating dendrite growth (Fig. 9c). This deposition mode represents an ideal benchmark in electrolyte design. Furthermore, the (002) crystal plane exhibits relatively low HER activity, reducing the likelihood of parasitic side reactions [143, 144]. Other crystal planes, such as Zn (103) and Zn (105), also demonstrate low HER activity, owing to their small angle (less than 30°) relative to the substrate surface, further supporting their potential in dendrite suppression strategies [145].Fig. 9a Chronoamperograms of Zn anodes under constant potential conditions [143]. b Comparison of nucleation overpotentials of Zn (100) and Zn (002) planes, derived from three independent symmetric cell experiments at varying current densities [143]. c Illustration of the hexagonal close-packed crystal structure of Zn [154]. d Top view of the geometrical configurations of H_2_O adsorbed on the Ti atom of SrTiO_3_ (110) plane [150]. Schematic diagram of the zinc deposition processes in e the conventional electrolyte and f the densified electrolyte [150]. Schematic illustration of Zn electrodeposition in g Bmim-containing and h Bmim-free electrolytes [154]
Constructing a Zn metal anode with a dominant (002) crystal plane orientation has emerged as a simple yet effective strategy for achieving high-performance AZIBs. However, conventional approaches such as etching and epitaxial electrodeposition often involve complex procedures and limited scalability [146–148]. Recent studies have demonstrated that specific electrolyte additives can effectively induce zinc deposition with preferred orientation origination by selectively adsorbing onto specific crystal planes. Inorganic additives, such as indium sulfate, tin oxide, and boric acid [143, 149], have been shown to regulate crystal orientation and inhibit the growth of dendrites. For instance, Deng et al. developed a densified aqueous electrolyte incorporating SrTiO_3_ metal oxide as an additive to achieve high-performance AZIBs [150]. DFT calculations revealed that Ti atoms on the SrTiO_3_ (110) plane exhibit high binding energy with water molecules, indicating a strong water affinity. Consequently, SrTiO_3_ particles adsorb water molecules, altering Zn^2+^ solvation structure and facilitating the incorporation of SO_4_^2−^ into the solvation shell (Fig. 9d). Meanwhile, SrTiO_3_ exhibits excellent affinity for Zn^2+^ ions, thereby guiding zinc deposition along the Zn (002) plane (Fig. 9e, f), enabling a Frank–Van der Merwe layer-by-layer growth mechanism. As a result, Zn||MnO_2_ cells assembled with this densified electrolyte delivered a high specific capacity of 328.2 mAh g^−1^ at 1 A g^−1^ after 500 cycles.
Organic additives, such as sulfonate anions, alcohols, and sugars, have also demonstrated the ability to tailor zinc texture by interacting with specific crystallographic planes [61, 96, 151, 152]. Certain polymers, owing to their specific functional groups, can preferentially adsorb onto the anode surface and promote uniform Zn^2+^deposition along the (002) crystal plane [153]. Ma et al. proposed a novel organic cation-assisted non-epitaxial electrodeposition strategy using 1-butyl-3-methylimidazolium cation (Bmim) as a paradigm additive [154]. Mechanistic studies indicated that Bmim^+^ cations selectively adsorb on the Zn (002) plane while simultaneously suppressing the growth rate of this plane, ultimately exposing and stabilizing the (002) orientation (Fig. 9g, h). The synergistic effect of the textured (002)Zn anode and the Bmim-containing electrolyte endows excellent cycling stability for over 350 h at 20 mAh cm^−2^ with a discharge depth of 72.6%.
New-Type of Electrolytes
In AZIBs, liquid electrolytes are widely adopted due to their high ionic conductivity. However, the application also introduces challenges, such as persistent side reactions and the instability at the solid–liquid interface, which limit further enhancements in battery performance [155, 156]. To overcome these limitations, hydrated eutectic electrolytes and gel electrolytes have emerged as promising alternatives for the next generation of AZIBs.
Hydrated Eutectic Electrolytes
Hydrated eutectic systems are formed by dissolving another crystallographically compatible salt or certain organic additives into hydrated salts, enabling the formation of eutectic mixtures with high salt concentrations and extended electrochemical stability windows. These systems can alter the Zn^2+^ solvation structure and reconstruct the interfacial phase of the EDL, thereby expanding the electrochemical stability window and enhancing ion transport [157, 158].
Under supersaturated conditions, ions disrupt the hydrogen-bond network of both coordinated and free water molecules, resulting in suppressed water activity, broader stability window, and improved ionic conductivity, all of which contribute to high-performance AZIBs [159, 160]. By tuning the composition, hydrated eutectic electrolytes significantly alter the solvation structure of Zn^2^⁺ and reinforce the hydrogen bonding, effectively immobilizing free water molecules. The synergistic effect between the optimized solvation and the strengthened hydrogen-bonding networks collectively imparts the electrolyte with enhanced capabilities to suppress side reactions and facilitate ion transport, thereby significantly improving the overall battery performance. Chen et al. developed a hydrated eutectic electrolyte based on ethylene glycol (EG), Zn(OTf)2, and a trace amount of H_2_O. By precisely controlling the water content, they minimized the number of active water molecules. In this system, EG and OTf⁻ anions cooperatively coordinate with Zn^2^⁺ ions via hydrogen bonds, establishing a thermodynamically stable solvation structure of [Zn(H_2_O)2(EG)2(OTf)2]. This architecture confines water activity and stabilizes the solvation sheath, resulting in a wide electrochemical stability window, low viscosity, and a high ion transference number [161]. More recently, Jiang et al. proposed a dual-salt high concentration electrolyte (15 mol kg^–1^ ZnCl_2_ + 10 mol kg^–1^ NH_4_NH_2_SO_3_), denoted as DS-HCE. The modulated solvation structure in DS-HCE delivers high ionic conductivity and ultra-low water activity. Compared to the conventional 30 M ZnCl_2_, DS-HCE achieves a broader electrochemical stability window. As a result, Zn||Zn symmetric cells using DS-HCE exhibit outstanding cycling stability, maintaining a lifespan of 2,200 h at 0.5 mA cm⁻^2^ and 0.5 mAh cm⁻^2^ [162].
Gel Electrolytes
Gel electrolytes, which combine the structural stability of solid-state electrolytes with the high ionic kinetics of liquid electrolytes, have also become a focal point of research in the field of AZIBs [163, 164]. Compared to conventional aqueous electrolytes, gel electrolytes typically feature a porous and three-dimensional network composed of functional chemical components that facilitate the creation of efficient ion transport channels and enable uniform Zn^2+^ deposition (Fig. 10a, b) [165, 166]. Recent studies have shown that polar functional groups, such as hydroxyl, carboxyl, and sulfonic groups, are widely present in gel electrolytes. These groups promote directional Zn^2+^ migration, enhance ionic conductivity, and reduce overpotential induced by concentration polarization. For instance, Zeng et al. introduced amphiphilic groups into a hydrogel electrolyte, enabling three-dimensional Zn^2+^ diffusion and guiding the formation of crystalline zinc deposits (Fig. 10c, d) [166]. Furthermore, charged functional groups (e.g., sulfonate and imidazole groups) can modulate the Zn^2+^ solvated structure and induce preferential growth along the Zn (002) crystal plane (Fig. 10e) [167]. The hydrophilic segments within gel electrolytes form hydrogen bonds with water molecules, effectively reducing the content of free water, thereby minimizing side reactions and providing a safer and more stable electrochemical environment [168–170]. Building on this strategy, He et al. incorporated a betaine-type zwitterionic monomer into Zn(ClO_4_)2 and copolymerized it with acrylamide to construct a gel framework with both hydrophilicity and charged groups (denoted as ADC-gel). The interaction between water and hydrophilic groups imparts high water retention, while the polymer backbone, under an applied electric field, forms directional ion migration channels along the aligned zwitterionic side chains, thereby enhancing Zn^2+^ transport efficiency (Fig. 10f). This gel electrolyte confers an extremely long cycle life on Zn||Zn symmetric batteries, with a cycle life exceeding 3,000 h at a current density of 0.5 mA cm^−2^ [171].Fig. 10a Schematic illustration of intermolecular interactions within the ADC-gel matrix [171]. b Schematic representation of ion migration channels formed in PAM-AAS-QCS hydrogel electrolyte [166]. Schematic diagrams comparing Zn plating behaviors in c conventional liquid electrolyte and d the PAM-AAS-QCS hydrogel electrolyte [166]. e Mechanism of Zn anode texture regulation using gel electrolyte [167]. f Schematic depiction of ion channel formation under the applied electric field [171]. Illustration of the adhesive mechanism of zwitterion hydrogel electrolyte at g high and h low water contents [175]. i Functional mechanism of zwitterion hydrogel electrolytes [175]. j Ionic transportation mechanism under lean-water conditions: including the formation of directional ion transportation channels and lubrication mechanism [175]
Despite these advancements, gel electrolytes still face challenges, including low ionic conductivity, limited mechanical strength, and poor environmental sustainability [172]. To address these issues, water-in-salt hydrogel electrolytes have been proposed [173, 174]. These systems suppress H_2_O activity, broaden the electrochemical stability window, and improve ionic conductivity [157]. Their low water content enhances interfacial adhesion, offering promise for flexible and wearable energy storage devices (Fig. 10g, h). For instance, Wang et al. reported a lean-water hydrogel electrolyte based on a polymeric zwitterion. The sulfonate groups exhibited both hydrophilic and zincophilic characteristics, facilitating Zn^2+^ transport. In this low-water environment, interactions between water molecules and charged groups result in the formation of molecular lubrication layers that reduce the interaction friction and enhance ion-pair mobility (Fig. 10i, j) [175]. Moreover, supramolecular design strategies integrating conductive polymers into flexible hydrogel matrices have been shown to improve conductivity, mechanical toughness, and self-healing capabilities. These multifunctional hydrogels enable the development of highly stretchable, biocompatible, and self-repairing wearable sensors [176, 177].
Commentary and Comparison
The growth of dendrite, HER, and surface passivation have profound impacts on the discharge capacity, Coulombic efficiency, and cycling stability of AZIBs. To address these issues, constructing a thermally stable metal anode–electrolyte interface is crucial [178, 179]. Among various approaches, including anode structural modification and the introduction of interfacial catalysts, electrolyte engineering has proven to be one of the most effective approaches [180–182]. Based on this, several feasible optimization directions in electrolyte engineering can be proposed:
- Zinc salt optimization: The selection of appropriate zinc salts can regulate electrolyte pH and chemical stability, thereby establishing a more favorable electrochemical environment. Additionally, concentration adjustment enables the expansion of the electrochemical stability window, contributing to enhanced AZIBs performance.
- Electrolyte additives: Incorporating various additives, such as alkali metal ions for electrostatic shielding, organic molecules with strong adsorption capability, desolvation agents, SEI film precursors, and dynamic interfacial regulators, can significantly improve Zn^2+^ deposition behavior and suppress side reactions. Notably, these mechanisms often act synergistically rather than independently [183]. For instance, adsorption not only shortens the Zn^2+^ two-dimensional diffusion time, increases nucleation site density, and lowers the nucleation energy barrier to promote uniform and directional Zn^2+^ deposition, but also facilitates rapid interfacial passivation to form a robust artificial SEI layer. Therefore, additive design should comprehensively consider the interaction among these multiple functional mechanisms.
- Development of advanced electrolytes: Emerging systems, including gel and hydrogel electrolytes with reduced free water content, offer enhanced stability and suppressed reactivity. These systems enable AZIBs to operate over a wide temperature range and at high current densities, making them particularly suitable for flexible and wearable electronic devices.
To aid comparison and provide clarity, Table 3 summarizes the electrolyte additives discussed in this review along with their corresponding electrochemical performance metrics. Table 3. Electrolyte additives and their impact on battery performanceAdditiveDeviceMain mechanismCycling performanceReferencesLiClZn||ZnSelf-healing electrostatic shield > 1000 cycles at 100 mA cm⁻^2^CE ≈ 99.9%[100]Rb_2_SO_4_Zn||VO_2_Self-healing electrostatic shield71.6% capacity retention after 500 cycles at 5 A g⁻^1^[105]Ga(NO_3_)3_Zn||VO_2_Self-healing electrostatic shield122.9 mAh g⁻^1^ retained after 1500 cycles at 5 A g⁻^1^[104]Graphene Oxide (GO)Zn||MnO_2_Adsorption effects93% capacity retention after 250 cycles at 1 A g⁻^1^[108]6-Aminohexanoic acid (6AA)Zn||MnO_2_Adsorption effects > 97.5% CE over 200 cycles at 0.5 A g⁻^1^[113]SDBS (anionic surfactant)Zn||MnO_2_Adsorption Effects85% capacity retention after 1000 cycles at 1 A g⁻^1^[109]Hyaluronic acid (HA)Zn||ZnDesolvation2200 h at 1 mA cm⁻^2^[121]Hyaluronic acid (HA)Zn||MnO_2_Desolvation61.4% retention after 1000 cycles at 1 A g⁻^1^[121]2-PropanolZn||ZnDesolvation500 h at 15 mA cm⁻^2^[120]Dodecyl sulfate (OSO_3_R⁻)Zn||N-doped porous carbonDesolvation89.1% retention after 40 000 cycles at 5 A g⁻^1^[122]APA (amphiphilic polymer)Zn||ZnDesolvation8800 h at 1 mA cm⁻^2^[123]PAA (polyacrylic acid)Zn||ZnDesolvation3293 h at 5 mA cm⁻^2^[124]Zn(H_2_PO_4)_2_Zn||V₂O₅SEI formationnearly 100% CE at 0.8 A g^−1^ over 1000 cycles[130]trimethylethyl ammonium trifluoromethanesulfonate (Me_3_EtNOTf)Zn||VOPO_4_SEI formationaverage CE of 99.9% at 2 A g^−1^ after 6000 cycles[131]Poly (N-vinylpyrrolidone-co-methyl acrylate) dipolymerZn||NVOSEI formationcapacity retention of 94.33% for 60 days[132]sucrose biomoleculeZn||NVOSEI formation73.1% capacity retention at 1 A g^−1^ over 670 cycles[133]N-[2-(3,4-dihydroxyphenyl)ethyl]-2-methylacrylamideZn||NVOSEI formationCE of 99.84% at 1 A g^−1^ after 1000 cycles[137]saccharinZn||MnO_2_SEI formationCE of 99.9% after 7500 cycles[138]positively charged chlorinated graphene quantum dotZn||LMOSEI formationcapacity retention of 86% after 100 cycles[139]graphitic carbon nitride quantum dotsZn||MnO_2_SEI formationnearly 100% CE over 500 cycles[15]SrTiO_3_Zn||MnO_2_Regulate crystal planespecific capacity of 238 mA h g^−1^ after 200 cycles at 0.5 A g^−1^[150]polyethylene glycolZn||V₂O₅Regulate crystal planecapacity retention of 84% at 15 A g^−1^ after 500 cycles[151]Dextran(with a 70 000 molecular weight)Zn||MnO_2_Regulate crystal planecapacity retention of 83% at 1 A g^−1^after 3000 cycles[152]2-methacryloyloxyethyl phosphorylcholine and N-acryloyl glycinamideZn||PANIRegulate crystal planecapacity retention of 90% at 15 A g^−1^ after 12,000 cycles[153]1-butyl-3-methylimidazolium cationZn||VOHRegulate crystal planecapacity retention of 90% at 0.2 A g^−1^ after 300 cycles[154]
It is particularly noteworthy that the aforementioned electrolyte engineering strategies must be considered in tandem with their potential effects on cathode materials. As summarized in Table 3, the compatibility between electrolyte additives and cathode materials plays a critical role in the overall electrochemical performance of AZIBs. For instance, vanadium oxide cathodes are susceptible to dissolution, prompting the use of Zn(CF_3_SO_3_)2-based electrolytes to mitigate degradation. Vanadate-based cathodes often require the incorporation of metal ions to suppress active material leaching. Manganese-based cathodes frequently employ mixed electrolyte systems to alleviate Mn dissolution. For Prussian blue analogs (PBAs), controlling electrolyte concentration or introducing metal ion additives (such as Ni^2+^) is essential to prevent structural phase transitions and capacity fading [58, 184]. Therefore, future research should focus on the rational design of cathode–anode compatible electrolyte systems to achieve coordinated optimization of full cell performance.
Summary and Prospects
This review highlights the electrolyte-related challenges associated with Zn metal anode in AZIBs and discusses corresponding strategies for their mitigation. Key issues, including dendrite growth, HER, and surface passivation, substantially compromise the electrochemical performance and safety of Zn metal anodes. Dendrite formation not only reduces the active anode surface area but also poses a risk of internal short circuits. Concurrently, HER and surface passivation induce energy loss and, in some cases, can cause battery swelling, posing serious safety concerns. The complexity and interconnectivity of these degradation mechanisms have prompted the development of diverse mitigation strategies, among which electrolyte structure engineering has emerged as one of the most effective.
Based on the above, viable approaches in electrolyte design include zinc salts optimization, the development of functional additives, and the design of novel electrolytes. However, several pressing challenges and emerging directions remain to be addressed:
- Dynamic Reconstruction Techniques: Conventional stationary interfacial modification methods often fail to maintain long-term efficacy in complex operating environments due to continuous microstructural evolution and localized electrochemical heterogeneity. A dynamic and adaptive interfacial layer capable of real-time self-regulation is essential. Recent developments in dynamic self-healing and dynamic interface reconstruction technologies have shown promise in stabilizing the anode/electrolyte interface via autonomous structural reconfiguration during charging/discharging processes.
- Artificial intelligence (AI)-Guided Additive Screening: The integration of AI enables high-throughput screening, property prediction, and intelligent materials discovery with minimal computational cost. Future research might focus on leveraging AI for efficient additive design and formulation optimization to meet specific requirements, such as reducing the nucleation barriers, controlling the nucleation rate, promoting Frank–Van der Merwe deposition, etc. These advancements will accelerate the development of next-generation AZIBs systems with enhanced interfacial control and electrochemical performance.
- Adaptability to Extreme Conditions: Under harsh operating conditions, such as low temperatures and high current densities, the kinetics of ion transport and interfacial processes become increasingly complex. Tailoring electrolytes to ensure high-rate capability and robust cycling stability under such conditions remains a key direction for future development.
- Application-specific Electrolyte Engineering: Beyond improving intrinsic performance, the electrolytes for AZIBs should be tailored for diverse application scenarios. Strategic electrolyte modifications can broaden AZIBs utility across fields, such as biodegradable electronics, implantable medical devices, and flexible/wearable systems, thereby facilitating cross-disciplinary innovation.
Despite considerable advancements in electrolyte structure regulation, unresolved challenges related to Zn anodes require more systematic and integrative solutions. Given the intrinsic coupling of dendrite growth, HER, and passivation, single-strategy approaches often face inherent limitations. Therefore, multi-functional and synergistic strategies that simultaneously improve performance, cost-efficiency, and scalability are urgently needed. In this context, a deeper understanding of interfacial mechanisms and ion transport kinetics is vital for enhancing electrochemical efficiency and prolonging battery lifespan. Furthermore, this review emphasizes the importance of developing electrolyte systems that are not only functionally robust and chemically stable but also environmentally sustainable and economically viable. Future research should incorporate comprehensive assessments of ecological impact and cost to guide the practical deployment of AZIBs. Given their potential in large-scale, sustainable energy storage, continued innovation in electrolyte structure design will be instrumental in positioning AZIBs as a leading solution for future clean energy technologies.