Distal Renal Tubular Acidosis With Sensorineural Deafness in a Saudi Female: A Case Report of an ATP6V1B1 Mutation in a Consanguineous Family
Abeer Alrasheed, Nouf Alyabis, Soud A Alrasheed, Reem Alrasheed

TL;DR
A Saudi girl with kidney acidification issues and hearing loss had a rare genetic mutation in ATP6V1B1, highlighting the importance of genetic testing in consanguineous families.
Contribution
Reports a novel ATP6V1B1 mutation (c.1037C>G; p.P346R) in a consanguineous family with dRTA and sensorineural deafness.
Findings
An 11-year-old Saudi girl was diagnosed with dRTA and sensorineural hearing loss due to a homozygous ATP6V1B1 mutation.
Parents were heterozygous carriers, indicating autosomal recessive inheritance.
The case emphasizes the need for early genetic diagnosis and multidisciplinary care in such conditions.
Abstract
Distal renal tubular acidosis (dRTA) with sensorineural deafness is a rare entity inherited in an autosomal recessive manner caused by mutations in the ATP6V1B1 gene leading to defective acidification function in the distal nephron, cochlea, and endolymphatic sac. We report the case of an 11-year-old Saudi girl with dRTA and congenital sensorineural hearing loss. Genetic testing revealed a homozygous mutation in the ATP6V1B1 gene (c.1037C>G; p.P346R). Both parents were heterozygous carriers. This case highlights the clinical and genetic features of dRTA in a consanguineous family and underscores the importance of early genetic diagnosis and multidisciplinary management.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
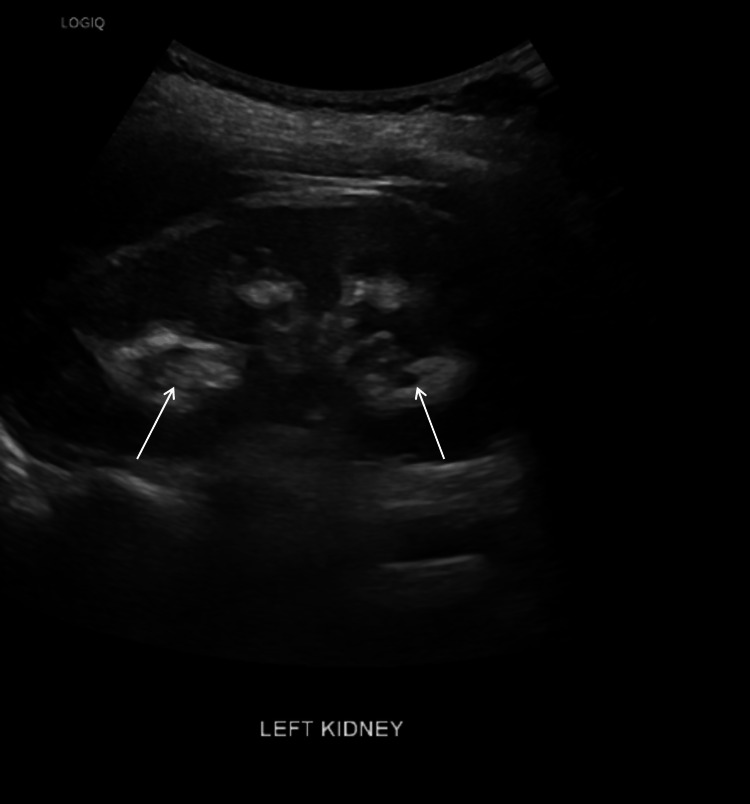 Figure 1
Figure 1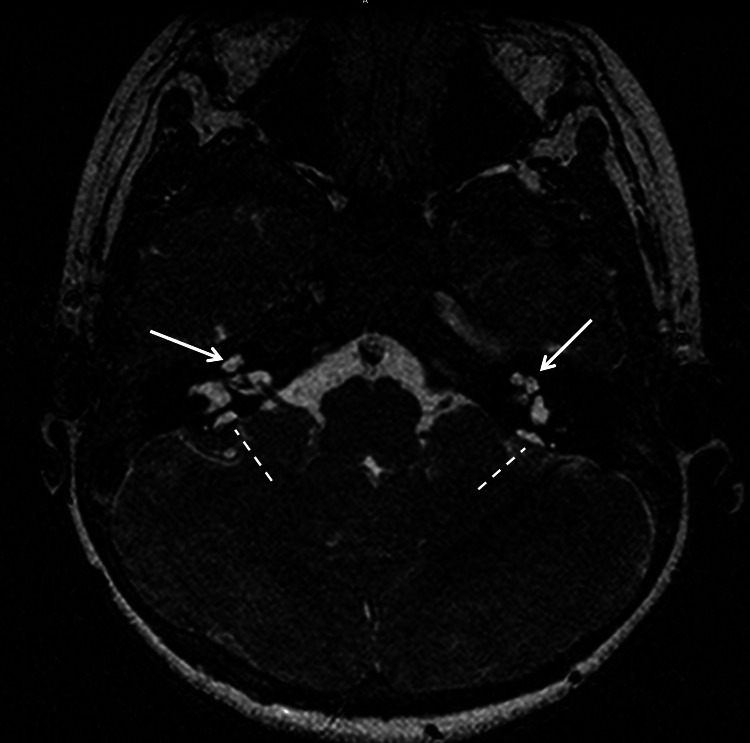 Figure 2
Figure 2| Parameters | Result | Reference Range | Units |
| Serum sodium | 136 | 136-145 | mmol/L |
| Serum potassium | 3.0 | 3.5-5.1 | mmol/L |
| Serum chloride | 114 | 98-107 | mmol/L |
| TCO₂ | 11 | 22-29 | mmol/L |
| Serum creatinine | 38 | 53-78 | µmol/L |
| Blood urea nitrogen | 5.4 | 3.0-7.5 | mmol/L |
| Serum calcium | 2.4 | 2.1-2.55 | mmol/L |
| Serum phosphorus | 1.43 | 0.81-1.45 | mmol/L |
| Serum albumin | 42 | 35-50 | g/L |
| Serum anion gap | 14 | 8-16 | mmol/L |
| Random urine sodium | 50 | >20 | mmol/L |
| Random urine potassium | 30 | 20-100 | mmol/L |
| Random urine random urine | 40 | 30-260 | mmol/L |
| Urine pH | 8.5 | 5.0-7.0 | — |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsIon Transport and Channel Regulation · Ion channel regulation and function · Hearing, Cochlea, Tinnitus, Genetics
Introduction
Distal renal tubular acidosis (dRTA) is a rare inherited disorder caused by the inability of the kidney to secrete hydrogen ions in the distal tubule. This causes metabolic acidosis with a normal anion gap [1]. In some patients with dRTA, there is sensorineural hearing loss [2]. The mode of inheritance is autosomal recessive. It is most frequently caused by mutations in the ATP6V1B1 or ATP6V0A4 genes. Mutations in the SLC4A1 gene cause a milder form of dRTA that follows an autosomal dominant inheritance and often presents in adulthood; therefore, few pediatric cases have been reported. The ATP6V1B1 gene encodes a subunit of the H+-ATPase pump [3]. The pump plays a role in both renal acidification and inner ear function [4]. Mutations in the gene disrupt both these processes and lead to renal and auditory manifestations [3]. The disorder is rare worldwide [1]. It is, however, more common in populations with a high rate of consanguineous marriages [5]. Consanguinity is culturally accepted and common in Saudi Arabia [5,6]. There have been limited reports from Saudi Arabia of this combined presentation. Early diagnosis and treatment are required to reduce complications. A genetic diagnosis can confirm a definitive diagnosis and direct family counseling [7,8]. We report an 11-year-old Saudi girl with dRTA and severe sensorineural hearing loss, born to first-degree cousins. Two of her older brothers were also affected. A homozygous ATP6V1B1 mutation was identified in the patient and her affected siblings, while both parents were found to be heterozygous carriers.
Case presentation
An 11-year-old girl was referred for evaluation of failure to thrive and hypokalemia. She was the sixth and youngest child born to a first-degree consanguineous couple. Two of her older brothers were known to have renal tubular acidosis (RTA) and deafness. She was born at 39 weeks of gestation, with a birth weight of 3.1 kg. She was found to have a hearing deficit since birth, which was confirmed using automated auditory brainstem response (AABR) testing.
On physical examination, she weighed 20.4 kg and measured 115 cm in height, both below the third percentile. She had mild bilateral genu valgum and profound hearing loss. The rest of the examination was normal. Investigations showed the following: laboratory results are summarized in Table 1. There was no pyuria, glycosuria, or hematuria. The urine anion gap was positive.
Renal ultrasound (Figure 1) revealed bilateral medullary nephrocalcinosis. Audiometry confirmed severe bilateral sensorineural hearing loss.
Renal ultrasound of the left kidney demonstrating hyperechoic medullary pyramids (arrows), consistent with medullary nephrocalcinosis
These findings confirm the diagnosis of distal RTA (dRTA) with sensorineural deafness. After obtaining consent from parents, genetic analysis was performed that showed a homozygous pathogenic variant in the ATP6V1B1 gene c.1037C>G; p.P346R.
Subsequently, genetic screening for the family members was carried out after obtaining consent. The results showed that both parents and one brother were heterozygous for the mutation, while the two brothers with RTA and deafness had the homozygous ATP6V1B1 gene mutation (c.1037C>G; p.P346R). The remaining two brothers were not affected.
Brain MRI revealed bilateral cochlear malformation consistent with Mondini's deformity and dilated vestibular aqueducts (right: 1.9 mm; left: 2.4 mm) (Figure 2). These structures communicated with small extra-axial fluid signals posterior to the mastoid bones, representing endolymphatic sacs. The vestibule and lateral semicircular canals were well developed and normally configured. The above-described findings were consistent with bilateral Mondini’s malformation and bilateral endolymphatic sacs.
MRI of the brain showing bilateral malformed cochleae (solid arrows) and endolymphatic sacs (dotted lines), both representing Mondini’s malformation
She was treated with Polycitra-K (citric acid/K-citrate/Na citrate) to provide 6 mmol/kg/day of bicarbonate equivalent. Aural rehabilitation with hearing aids and speech therapy was initiated, and she was referred to an otolaryngologist for consideration of a cochlear implant to support her language development.
Discussion
Acid-base homeostasis is critical for normal cellular function, growth, and development. The kidneys play an important role by reabsorbing bicarbonate, mainly in the proximal tubules, and by secreting acid in the distal tubules and collecting ducts of the nephrons. The net acid secretion is mediated largely by an energy-dependent proton pump (H+-ATPase) located in the apical membrane of α-intercalated cells of the distal nephron [9,10]. Failure of the urine acidification process results in dRTA.
In families with dRTA and early-onset hearing loss, the putative mutation was identified in the ATP6V1B1 gene encoding the B subunit of the H+-ATPase pump located in the apical surface of the α-intercalated cells in the distal tubule and is also expressed in the human cochlea and endolymphatic sac epithelium. An active acidification process constantly occurs in the ear to maintain endolymph pH near 7.4 in the cochlea and closer to 6.6 in the endolymphatic sac. Failure of this process leads to sensorineural deafness [11]. The mutation identified in our patient and her older brothers explains the occurrence of dRTA and early-onset deafness. Mutations in ATP6V1B1 were first identified as a cause of dRTA and deafness by Karet et al. in 1999 [6]. Since then, several variants of these gene mutations have been identified in affected individuals from various populations worldwide, including Saudi Arabia [12]. Most reports indicate its occurrence in consanguineous families and describe delayed diagnosis because of overlapping symptoms with more common pediatric conditions such as rickets and dehydration [7]. Consanguineous marriage is socially acceptable in Saudi Arabia [8]. The total consanguinity rate is approximately 57.7%, leading to a higher prevalence of autosomal recessive diseases like dRTA and deafness [8]. The current report of three siblings and ATP6V1B1 mutations illustrates this risk.
Gene testing based on sequencing of exon 12 at the ATP6V1B1 gene for the whole family members of the affected individual helps in the diagnosis and identification of carriers and forms the basis for counseling and clinical management [13]. The main treatment of dRTA is alkali therapy to correct metabolic acidosis and prevent some complications such as growth failure, kidney stones, and renal failure [3,14]. Alkali therapy does not alter the course of the sensorineural deafness. Management of such cases is complex and requires long-term follow-up by a multidisciplinary team, consisting of a pediatric nephrologist, otolaryngologist, urologist, genetic counselor, and speech therapist [15].
Conclusions
This case report describes three siblings from a Saudi consanguineous family with dRTA, sensorineural deafness, and ATP6V1B1 gene mutation. Imaging confirmed Mondini's malformation and dilated vestibular aqueducts. Early diagnosis and genetic confirmation are essential for appropriate management and family counseling. A multidisciplinary care plan is necessary to address both renal and auditory complications of this rare condition.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Clinical and molecular findings in three Moroccan families with distal renal tubular acidosis and deafness: report of a novel mutation of ATP 6V 1B 1 gene Curr Res Transl Med Boualla L Jdioui W Soulami K Ratbi I Sefiani A 586420162714059310.1016/j.retram.2016.01.005 · doi ↗ · pubmed ↗
- 2ATP 6V 1B 1 mutations in distal renal tubular acidosis and sensorineural hearing loss: clinical and genetic spectrum of five families Ren Fail Uzak AS Cakar N Comak E Yalcinkaya F Tekin M 128112843520132392398110.3109/0886022 X.2013.824362 PMC 5483946 · doi ↗ · pubmed ↗
- 3Distal renal tubular acidosis. Clinical manifestations in patients with different underlying gene mutations Pediatr Nephrol Alonso-Varela M Gil-Peña H Coto E Gómez J Rodríguez J Rodríguez-Rubio E Santos F 152315293320182972577110.1007/s 00467-018-3965-8 · doi ↗ · pubmed ↗
- 4Importance of early audiologic assessment in distal renal tubular acidosis Int Med Case Rep J Swayamprakasam AP Stover E Norgett E Blake-Palmer KG Cunningham MJ Karet FE 711420112375489710.2147/IMCRJ.S 13667 PMC 3658229 · doi ↗ · pubmed ↗
- 5Distal renal tubular acidosis and its relationship with hearing loss in children: preliminary report Iran J Kidney Dis Sharifian M Esfandiar N Mazaheri S 20220642010 https://ijkd.org/index.php/ijkd/article/view/188/19920622307 · pubmed ↗
- 6Mutations in the gene encoding B 1 subunit of H+-AT Pase cause renal tubular acidosis with sensorineural deafness Nat Genet Karet FE Finberg KE Nelson RD 8490211999991679610.1038/5022 · doi ↗ · pubmed ↗
- 7Genetic studies in a family with distal renal tubular acidosis and sensorineural deafness Indian Pediatr Sethi SK Singh N Gil H Bagga A 425427462009 https://www.indianpediatrics.net/may 2009/425.pdf 19478356 · pubmed ↗
- 8Hearing impairment in association with distal renal tubular acidosis among Saudi children J Laryngol Otol Zakzouk SM Sobki SH Mansour F al Anazy FH 9309341091995749994310.1017/s 0022215100131706 · doi ↗ · pubmed ↗