Draft genome sequence data of Colletotrichum siamense isolated from Camellia japonica in the United States
Kenneth R. Leep, Renee S. Arias, Warren E. Copes, Siva P. Kumpatla

TL;DR
Researchers sequenced the genome of Colletotrichum siamense, a damaging fungus found on Camellia japonica in the U.S., for the first time.
Contribution
This is the first report of C. siamense genome sequences from ornamental plants in the United States.
Findings
C. siamense was isolated from Camellia japonica in Mississippi and sequenced.
Genome data were submitted to NCBI with accession numbers SAMN48929844 and SAMN49025292.
The data will help develop diagnostics and understand the pathogen's diversity and spread.
Abstract
Colletotrichum siamense Prihastuti, L. Cai & K.D. Hyde is an economically important fungal pathogen that causes damage to diverse horticultural and agronomic crops including fruits, vegetables, and ornamentals. While its prevalence on, and damage to ornamental crops has been reported in many tropical and subtropical countries, to date it was not documented on ornamentals in the United States. Here, we report the isolation of C. siamense from Camellia japonica L. in the United States and the first genome sequence of this pathogen. C. siamense isolates GCC05 and GCC08 were obtained from C. japonica plants in George County, Mississippi. Whole genome sequencing of GCC05 and GCC08 was performed as paired-end reads of 150 bp using NovaSeqXPlus. Clean reads were de novo assembled and were also mapped to C. siamense reference genome sequence, Cg363, and genome-wide variants were analyzed.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
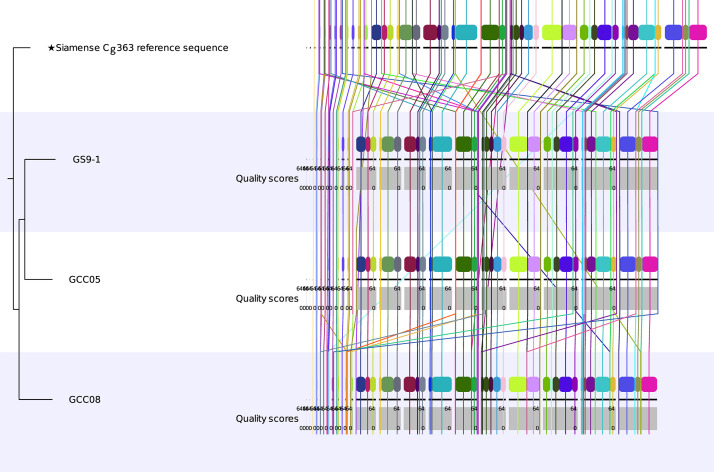 Figure 1
Figure 1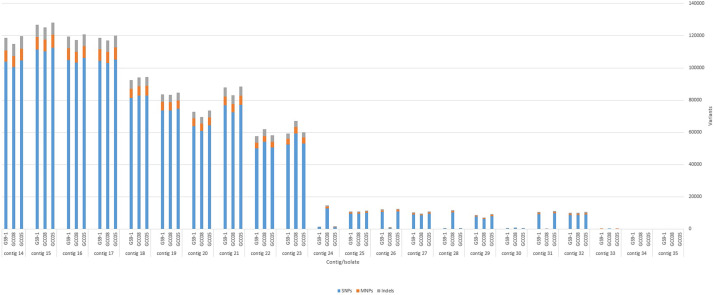 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant Pathogens and Fungal Diseases · Fungal Plant Pathogen Control · Mycorrhizal Fungi and Plant Interactions
Specifications TableSubjectBiologySpecific subject areaFungal plant pathogen genomicsType of dataGenome sequences, raw data, De novo assembly, filtered and trimmed reads, reference mapping data, tables, figuresData collectionColletotrichum siamense was isolated from symptomatic Camellia japonica plants in George County, Mississippi, USA, in 2023. The whole genome was sequenced on Illumina NovaSeqXPlus platform at the UC Davis Genome Center, California. Data were processed using CLC Genomics Workbench v25.0.1. Clean reads >120 nucleotides were de novo assembled, and contigs were used to generate alignments for phylogenetic identification. Clean reads were also mapped to the reference genome Cg363 and variants (SNPs, MNPs, and Indels) were identified.Data source locationC. siamense was collected from George County, Mississippi, USA, 30° 51′ 54.5436′′ N, 88° 32′ 43.2054″ W. Sequencing data were placed in public repository: NCBI.Data accessibilityBioProject PRJNA1273064, accession numbers SAMN48929844 and SAMN49025292. Repository name: National Center for Biotechnology Information (NCBI). Direct URL to data:https://dataview.ncbi.nlm.nih.gov/object/PRJNA1273064?reviewer=6so5e1rsfphvgqpvouimi3l034
Value of the Data
1
- •This is the first time Colletotrichum siamense has been isolated from an ornamental plant, Camellia japonica, in the United States.
- •This is the first report of whole genome sequencing of C. siamense on ornamental plants in the United States. Given the phenotypic plasticity of Colletotrichum [1], the availability of the genomes reported here will allow unambiguous identification of this pathogen in the region.
- •The ornamental horticulture industry is a significant economic contributor in the specialty crops category in the United States. Identification and management of new fungal diseases is critical for reducing the damage to crops and thereby economic losses.
- •The C. siamense genomes reported here show many genetic variants when compared to the reference genome sequence of Cg363 from China. While a large number of single- and multi-nucleotide polymorphisms (SNPs, MNPs) and Insertion/Deletions (Indels) were observed throughout the genome, SNPs were found to be the major category of variants. The genome information is critical to understand the unique and differentiating features of the isolates found in Mississippi, in the United States, compared to C. siamense strains predominantly reported from tropical and subtropical countries elsewhere.
- •Mining and comparative analysis of C. siamense genome reported here and other genomes enable development of molecular markers which in turn can facilitate analysis of population structure, improve the resolution of phylogenetic trees, and identify pathogen strains to which plant cultivars can show resistance or susceptibility.
- •The data reported here will be valuable for the development of diagnostics for accurate identification, tracking, and management of this pathogen on ornamentals as well as on other horticultural and agronomic crops in the United States.
- •The genomic data provided here are also of substantial importance for studies on the evolution of the species and in understanding the genomic basis for broad or narrow host range of C. siamense pathogens.
Background
2
The Colletotrichum genus includes many important species that cause anthracnose on a wide range of host plants worldwide leading to significant economic losses, and consequently it is considered one of the top ten most damaging plant pathogens [2]. Recent diversity and phylogenomic analyses divided 280 Colletotrichum species into 16 species complexes and 15 singletons [3]. The Colletotrichum gloeosporioides species complex (CGSC) is one of the most devastating phytopathogenic species complexes and its members cause damage to diverse horticultural and agronomic crops [2,4]. C. siamense, a member of the CGSC, causes damage to diverse plant species including fruits, vegetables, and ornamentals in several tropical and subtropical countries [5,6].
In the United States, ornamental horticulture is a significant economic contributor, accounting for a third of the total value of specialty crops and 10 % of the value of total crop production [7]. In addition to managing existing diseases, proactively identifying, tracking, and managing new pathogens is important for maintaining the productivity of the ornamental industry. While some species of Colletotrichum have been reported on ornamental plants in the United States, to our knowledge, C. siamense has not been documented as a pathogen. Here, we provide the first report of C. siamense on C. japonica, the most important ornamental Camellia in the United States.
Accurate identification of fungal species is crucial for understanding etiology and pathogenicity, exploring host range, creating effective disease management strategies, and developing diagnostics for tracking pathogen spread [8]. Internal transcribed spacer (ITS) sequences by themselves or in combination with multi-locus concatenated sequences in phylogenetic analyses were successfully used to distinguish several Colletotrichum species [5]. However, even the resolution of most commonly used ITS was found to be insufficient to reliably differentiate C. siamense from several Colletotrichum species, thus requiring the use of specific genes or alternate approaches in those situations [9]. The availability of whole genome sequences of C. siamense and their comparison to the non-redundant reference sequences and other genomes enable differentiation of C. siamense from other species with high accuracy.
While C. siamense has been found on many hosts around the world, to date most reported cases of this species on ornamentals are from Asian countries along with reports from a few other countries that include Australia, Brazil, and Mexico. By identifying C. siamense, for the first time, in an ornamental crop in the United States this study extends the geographic range of C. siamense on ornamental crops. Most importantly, the genomic sequences provided here aid in the development of diagnostics for the identification, tracking, and management of this pathogen in the United States on horticultural as well as agronomic crops. The purpose of this research was to obtain the draft genome sequences of two C. siamense isolates, GCC05 and GCC08, from the United States and derive information on their similarity/dissimilarity to existing C. siamense genomes reported from other regions.
Data Description
3
Draft genome sequences for isolates GCC05 and GCC08 are available at the National Center for Biotechnology Information (NCBI) under the BioProject Number PRJNA1273064 with the accession numbers SAMN48929844 and SAMN49025292, respectively. The summary description of sequencing data is shown in Table 1. A third isolate, GS9–1, was also sequenced and initially included in the data and analyses, but it was later determined to be genetically identical to GCC05 (Fig. 1) and was, therefore, excluded from subsequent analyses and final reporting.Table 1. Genomic features and de novo assembly statistics for Colletotrichum siamense isolates GCC05 and GCC08.Table 1. AttributeGCC05GCC08Genome size (bp)57,743,33557,651,220Number of contigs165249Largest contig (bp)3,257,7443,380,721Average contig length (bp)264,878174,701Sequencing Coverage170.5x99.9xNumber of scaffolds116201N25 (bp)2,249,2271,275,315N50 (bp)1,050,410789,599N75 (bp)501,940384,783Overall GC Content52.37 %52.47 %Fig. 1. Whole genome alignment of Colletotrichum siamense isolates GCC05, GCC08, and GS9–1 to reference genome Cg363.Fig 1
The whole genome sequences of the isolates were aligned to the annotated reference genome sequence of isolate Cg363 (Biosample ID: SAMN09531620) [10]. The alignment results are presented in Fig. 1. Genome-wide variant analysis was performed based on the comparison of sequencing data of the isolates and the reference genome sequence (Fig. 2). Contigs 14 to 23 shown in Fig. 2 correspond to ten core chromosomes expected in the C. siamense genome [11]. Additional work needs to be done to determine whether one or more of the smaller contigs merge with the larger contigs or correspond to mini-chromosomes commonly observed in several species of the CGSC, including C. siamense [11].Fig. 2. Contig-wise distribution of variants. SNPs: single nucleotide polymorphisms; MNPs: multiple nucleotide polymorphisms; and Indels: Insertions and deletions.Fig 2
Experimental Design, Materials and Methods
4
Fungal isolation
4.1
Plant samples from Camellia japonica ‘Gunsmoke’ that exhibited foliar anthracnose symptoms and stem dieback were collected in George County, Mississippi, USA. Samples were surface sterilized in 10 % (v/v) sodium hypochlorite solution (1:10 dilution of commercial bleach, approx 5–9 % NaOCl), rinsed in ultra-pure sterile reverse osmosis water, and the healthy and diseased tissue margin of foliar and stem canker symptoms were placed on potato dextrose agar (PDA). Fungal isolates were recovered and pure isolates obtained by passaging mycelial tips on PDA. Resulting isolates were grown at 24 °C for 3–5 days under constant light and morphologically identified to the genus level (Colletotrichum) through microscopic examination of conidia and conidiophores from acervuli. Mycelial tips were extracted and grown in potato dextrose broth on a shaker for an additional 3–5 days at 24 °C in low light conditions. The resulting mycelial globules were removed and vacuum filtered to eliminate the supernatant broth. Globules were split open, and agar blocks extracted using a fire-sterilized scalpel. Globules were placed into sterile 5 ml centrifuge tubes and frozen at −80 °C overnight and subsequently placed in a freeze dryer for approximately 48 h. The lyophilized tissue was placed in a 2 ml bead beater vial approximately ¼ full of 1 mm zirconium silica beads, with 1 ml DEPC-treated, molecular biology grade water added. The vials were placed on a Biospec Products Mini-Beadbeater and beaten at 2600 rpm for 3 min at room temperature (22 ± 3 °C). (BioSpec Products, Inc, Bartlesville, OK, USA).
Isolates were deposited in the Agricultural Research Service Culture Collection at the Northern Regional Research Laboratory (NRRL) located in Peoria, IL, USA. These isolates were assigned NRRL numbers 64915 (GCC05) and 64917 (GCC08).
Genomic DNA preparation
4.2
DNA extraction for all three samples (GCC05, GCC08, and GS9–1) was initially performed using the Dynabeads DNA Direct Universal Kit (Fisher Scientific, USA) according to the manufacturer’s instructions; however, due to issues with impurities in GCC05, DNA was re-extracted from GCC05 using the Wizard Genomic DNA Purification Kit (Promega Corp, USA), again following the manufacturer’s instructions. The concentration of DNA was measured using a NanoDrop 1000 Spectrophotometer (Thermo Scientific, USA).
Genome sequencing and assembly
4.3
Microbial whole genome library (350 bp) was prepared using AB clonal Rapid Plus DNA kit. Sequencing was performed as paired-end reads of 150 base pairs using the Illumina NovaSeqXPlus platform by Novogene, at the UC Davis Genome Center (Davis, CA, USA). Genome sequencing produced a total of 43,535,962 raw reads for GCC05 and 34,337,994 raw reads for GCC08.
To generate clean reads, adapters P5 and P7 (see Table 2 for adapter sequences) were removed from the reads, and low-quality or short reads (<140 bp) were trimmed using CLC Genomics Workbench v25.0.2 (Qiagen, Aarhus, Denmark). The resultant clean reads were mapped to the reference genome of C. siamense (Strain ID: Cg363). De novo assembly was performed using the default settings in CLC Genomics Workbench v25.0.2.Table 2. Adapters used by Novogene sequencing.Table 2. AdapterSequenceP5AATGATACGGCGACCACCGAGATCTACAC[i5]ACACTCTTTCCCTACACGACGCTCTTCCGATCTP7GATCGGAAGAGCACACGTCTGAACTCCAGTCAC[i7]ATCTCGTATGCCGTCTTCTGCTTG
Genome completeness was assessed with Busco v6.0.0. Samples were compared against the Glomerellales database (glomerellales_odb12). GCC05 was assessed to have 99.3 % completeness (C:99.3 %, S:99.1 %, D:0.2 %, F:0.1 %, M:0.6 %, n:6103) while GCC08 was assessed to have 99.4 % completeness (C:99.4 %, S:99.2 %, D:0.2 %, F:0.1 %, M:0.5 %, n:6103) where C = complete Buscos, S = Single copy Buscos, D = Duplicate Buscos, F = Fragmented Buscos, and M = Missing Buscos.
Furthermore, genomes were scanned for contaminants using Kraken2 v2.14. Three significant contaminants were found: Homo sapiens, with 304 reads from GCC05 and 548 reads from GCC08; Cutibacterium acnes with 694 reads from GCC08 (no contamination in GCC05); and Malassezia restricta with 125 reads from GCC08 (no contamination in GCC05). All of these contaminant reads were removed from the raw reads before de novo assembly was performed.
Limitations
Not applicable
Ethics Statement
The current work does not involve human subjects, animal experiments, or any data collected from social media platforms.
CRediT Author Statement
Kenneth R. Leep: Conceptualization**,** Methodology, Formal analysis, Data curation, Writing – original draft, Writing – review & editing; Renee S. Arias: Conceptualization, Methodology, Resources, Formal analysis, Writing – review & editing; Warren E. Copes: Conceptualization, Funding acquisition, Resources, Writing – review & editing; Siva P. Kumpatla: Conceptualization, Methodology, Funding acquisition, Writing – original draft, Writing – review & editing.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Menicucci A.Iacono S.Ramos M.Fiorenzani C.Peres N.A.Timmer L.W.Prodi A.Baroncelli R.Can whole genome sequencing resolve taxonomic ambiguities in fungi?, the case study of Colletotrichum associated with ferns Front. Fungal Biol.62025154046910.3389/ffunb.2025.1540469 PMC 1190668540093768 · doi ↗ · pubmed ↗
- 2Dean R.Van Kan J.A.L.Pretorius Z.A.Hammond-Kosack K.E.Di Pietro A.Spanu P.D.Rudd J.J.Dickman M.Kahmann R.Ellis J.Foster G.D.The top 10 fungal pathogens in molecular plant pathology Mol. Plant Pathol.13201241443010.1111/j.1364-3703.2011.00783.x 22471698 PMC 6638784 · doi ↗ · pubmed ↗
- 3Liu F.Ma Z.Y.Hou L.W.Diao Y.Z.Wu W.P.Damm U.Song S.Cai L.Updating species diversity of Colletotrichum, with a phylogenomic overview Stud. Mycol.101202215610.3114/sim.2022.101.0136059896 PMC 9365046 · doi ↗ · pubmed ↗
- 4Ma Z.Liu F.Tsui C.K.M.Cai L.Phylogenomics and adaptive evolution of the Colletotrichum gloeosporioides species complex Commun. Biol.8202359310.1038/s 42003-025-08024-9PMC 1198236640204844 · doi ↗ · pubmed ↗
- 5Weir B.S.Johnson P.R.Damm U.The Colletotrichum gloeosporioides species complex Stud. Mycol.73201211518010.3114/sim 001123136459 PMC 3458417 · doi ↗ · pubmed ↗
- 6Udayanga D.Manamgoda D.S.Liu X.Chukeatirote E.Hyde K.D.What are the common anthracnose pathogens of tropical fruits?Fungal. Divers.61201316517910.1007/s 13225-013-0257-2 · doi ↗
- 7Wei X.Khachatryan H.Hodges A.Hall C.Palma M.Torres A.Brumfield R.Exploring market choices in the US ornamental horticulture industry Agribus 3920236510910.1002/agr.21769 · doi ↗
- 8Bhunjun C.S.Phillips A.J.S.Jayawardena R.S.Promputtha I.Hyde K.D.Importance of molecular data to identify fungal plant pathogens and guidelines for pathogenicity testing based on Koch’s postulates Pathogens.102021109610.3390/pathogens 1009109634578129 PMC 8465164 · doi ↗ · pubmed ↗