Exploring Shootin1’s oncogenic role within FGFR2 gene fusions
Volkan ERGİN, Mutlu ERDOĞAN, Ekrem YAŞAR, Sika ZHENG

TL;DR
This study identifies a new cancer-causing gene fusion involving FGFR2 and Shootin1, revealing how it drives tumor growth, especially in cholangiocarcinoma.
Contribution
The first molecular characterization of the FGFR2::SHTN1 fusion and its oncogenic mechanism via constitutive activation.
Findings
FGFR2::SHTN1 is an in-frame fusion retaining the FGFR2 tyrosine kinase domain.
Shootin1's coiled-coil domains mediate ligand-independent dimerization and FGFR2 activation.
The fusion represents a potent oncogenic driver in cancers like cholangiocarcinoma.
Abstract
Fibroblast Growth Factor Receptor (FGFR) gene fusions are recognized as pivotal oncogenic drivers, contributing to cancer initiation and progression across diverse malignancies. These fusions often represent significant therapeutic targets, particularly in challenging malignancies like cholangiocarcinoma. This study aimed to characterize the novel FGFR2::SHTN1 fusion, identify it as a de novo chimeric protein, and elucidate its precise oncogenic mechanism. FGFR2::SHTN1 fusions were identified via cancer genomics databases and modeled using AlphaFold and HADDOCK. SHTN1 variants were expressed in Neuro-2a cells for coimmunoprecipitation, purification, and native polyacrylamide gel electrophoresis to assess oligomerization. Structural modeling included membrane embedding with Chemistry at HARvard Macromolecular Mechanics–Graphical User Interface (CHARMM–GUI). We found that FGFR2::SHTN1…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
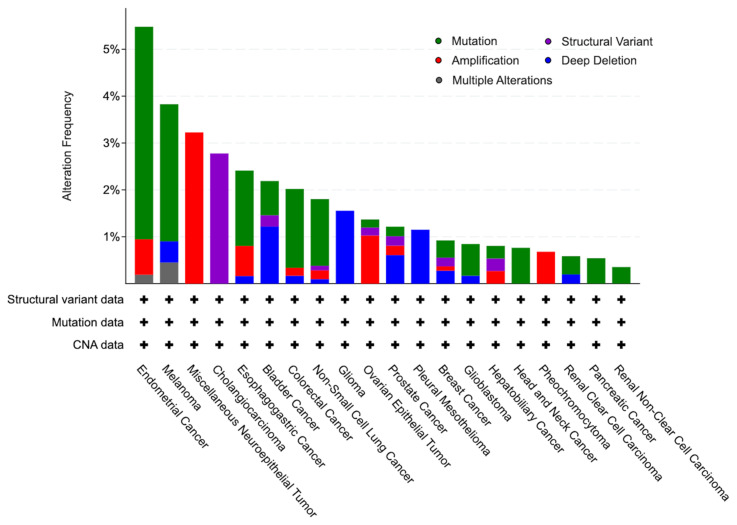 Figure 1
Figure 1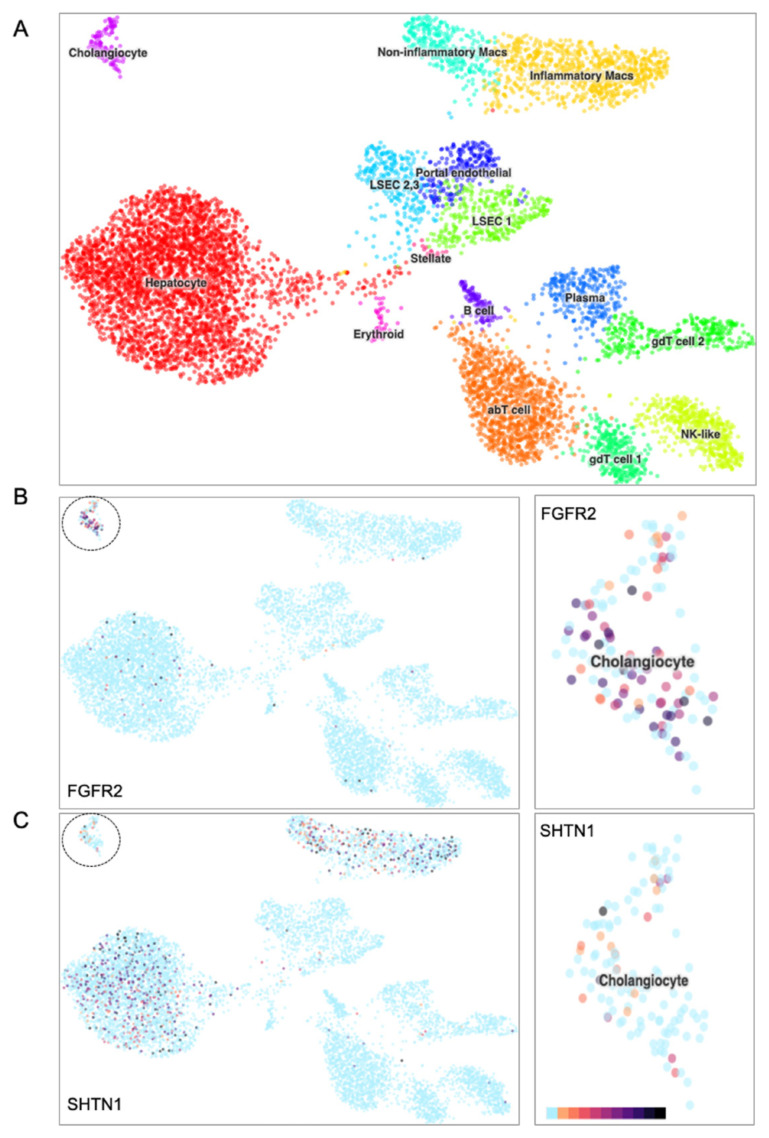 Figure 2
Figure 2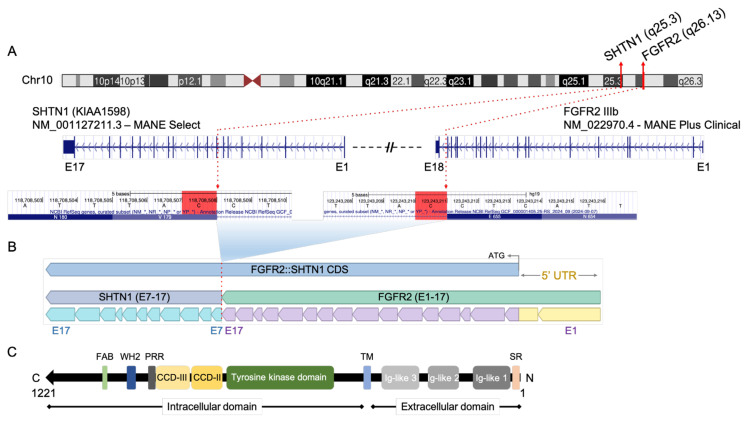 Figure 3
Figure 3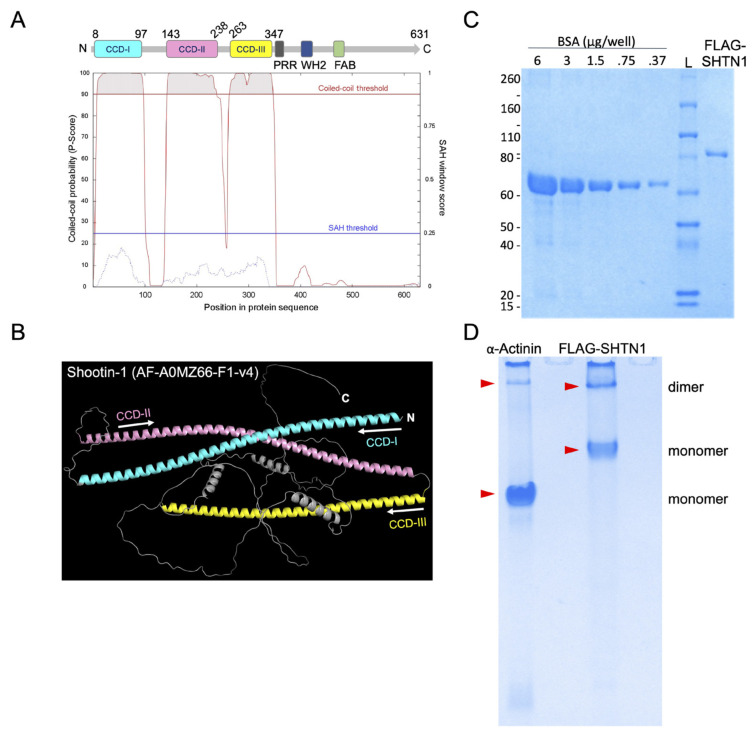 Figure 4
Figure 4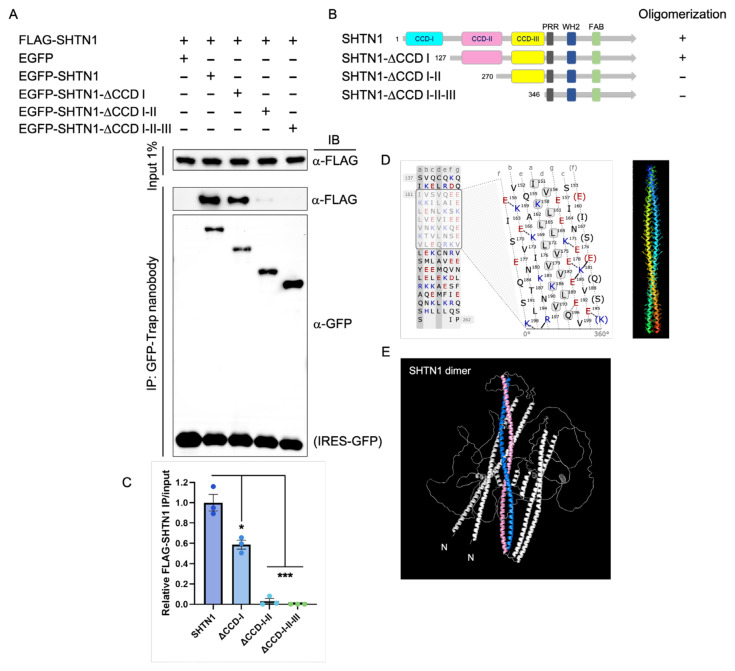 Figure 5
Figure 5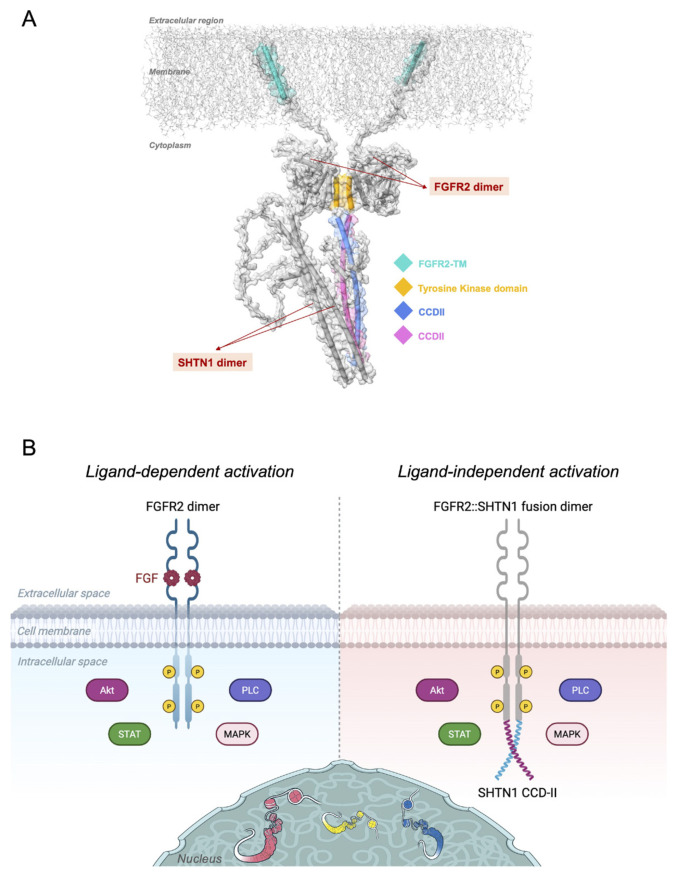 Figure 6
Figure 6- —National Institutes of Health (NIH)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFibroblast Growth Factor Research · IgG4-Related and Inflammatory Diseases · Chromatin Remodeling and Cancer
Introduction
SHTN1 (also known as Shootin1 or KIAA1598) is an intriguing protein initially identified for its critical roles in neuronal development. We previously demonstrated that it plays a central role in regulating neuronal polarity and axonogenesis by directly modulating cytoskeletal organization, a function tightly controlled by alternative splicing (Zhang et al., 2019; Ergin and Zheng, 2020).
In an earlier study, we showed that the alternative splicing of Shootin1 pre-mRNA generates a long isoform (SHTN1L) and a short isoform (SHTN1S) (Ergin et al., 2015). While SHTN1S is considered a neuron-specific isoform, SHTN1L is more broadly expressed across various tissues, including the brain (Higashiguchi et al., 2016; Zhang et al., 2019). However, the functional role of the Shootin1 protein in nonneuronal tissues remains poorly understood. In the neuronal context, SHTN1S promotes axon specification by asymmetrically accumulating in a single neurite, whereas the long isoform, SHTN1L, regulates cytoskeletal dynamics to control axon elongation during brain development (Zhang et al., 2019; Zheng, 2020).
Although both isoforms share an identical N-terminal sequence containing coiled–coil domains (CCD), the unique C terminus of SHTN1L grants it a distinct actin-binding property (Zhang et al., 2019; Ergin and Zheng, 2020). The difference in actin binding between the isoforms is a result of the presence or absence of the C-terminal sequence, which modulates the intramolecular interactions of their shared CCD parts. This functional multimodality arises from differential protein structures generated by alternative splicing. Note that, throughout this study, the protein name “Shootin1 or SHTN1” specifically refers to the long isoform, SHTN1L, unless otherwise noted.
Interestingly, alongside its function in cytoskeletal reorganization, Shootin1 has recently been implicated in the initiation and progression of various cancers, including bladder, lung, breast, and brain (Uguen and De Braekeleer, 2016; Qin et al., 2019; Li et al., 2020; Nicolò et al., 2023; Morin et al., 2024; Wang et al., 2024). It has been particularly identified and verified in oncogene fusions in cholangiocarcinoma (CCA) tumors (Krook et al., 2020; Li et al., 2020; Neumann et al., 2022; Silverman et al., 2022). The apparent dichotomy in Shootin1’s distinct biological roles suggests that the molecular mechanisms underlying its function may be coopted or dysregulated in oncogenic processes.
While the exact mechanisms underlying Shootin1’s involvement in tumorigenesis remain to be revealed, emerging evidence suggests its involvement in oncogenic gene fusions. In this regard, a significantly prevalent oncogenic partner of Shootin1 in such observed fusions is Fibroblast Growth Factor Receptor 2 (FGFR2) (Krook et al., 2020; Li et al., 2020; Silverman et al., 2022; Wang et al., 2024). It is well-established that most FGFR2 fusion partners promote the constitutive activation of FGF receptor signaling. These fusions typically involve the partner of the FGFR2 providing specific domains such as coiled–coil, SAM (sterile alpha motif), or caspase that facilitate persistent receptor dimerization, leading to ligand-independent activation of the kinase domain of FGFR2 (Parker et al., 2014; Tuna et al., 2019; De Luca et al., 2020).
The presence of three consecutive coiled–coil domains within the Shootin1 protein structure inherently suggests the potential for self-association via oligomerization (Ergin and Zheng, 2020). Taken together with the mechanisms governing FGFR2 oncogenic fusions, wherein coiled–coil domains mediate constitutive receptor dimerization, it is plausible that Shootin1 has the capacity for CCD oligomerization, which could likely be the mechanism driving FGFR2’s observed oncogenic activity.
The potential for CCD-mediated oligomerization in Shootin1, coupled with the notable genomic proximity and architecture of the SHTN1 and FGFR2 genes on human chromosome 10, suggests a compelling model for the observed propensity of these proteins to participate in oncogenic fusions. We hypothesized that a chromosomal deletion event between FGFR2 and SHTN1 could juxtapose the kinase domain of FGFR2 with the CCD domains of Shootin1. This juxtaposition would cause ligand-independent constitutive activation of FGFR2 signaling via Shootin1-induced dimerization, thereby driving oncogenesis, particularly in tissues where the FGFR2 promoter is active. In this study, we propose a mechanistic model to elucidate the plausibility of the FGFR2::Shootin1 oncogene fusion by providing critical evidence for Shootin1 oligomerization and establishing a link between constitutive FGFR2 activity across various forms of malignancy.
Materials and methods
2.1. Databases and computational tools
Alterations in the SHTN1 gene across various cancer types were visualized using cBioPortal1 (Gao et al., 2013). FGFR2::SHTN1 fusion events were identified using cBioPortal and the COSMIC database2 (Tate et al., 2019). Genomic coordinates of in-frame breakpoints in FGFR2 and SHTN1 were obtained from the UCSC Genome Browser on the human genome assembly GRCh37/hg19. Exon boundaries for each gene were retrieved from the Consensus CDS (CCDS) database23 (Pruitt et al., 2009). Domain compositions of the wild-type genes and putative fusion products were defined through literature review (Ross et al., 2014; Helsten et al., 2016; Zheng, 2020; Brown et al., 2023). Liver tissue single-cell RNA-seq datasets were accessed via the UCSC Cell Browser interface4.
The presence of a coiled–coil domain in Shootin1 was confirmed using the Waggawagga server5 (Simm et al., 2014), which employs the ab initio coiled–coil prediction algorithm MARCOIL (Delorenzi and Speed, 2002). The computationally predicted coiled–coil domain II (CCD-II), implicated in oligomerization, was modeled using CCBuilder 2.06 (Wood and Woolfson, 2018). To provide a structural basis for the putative fusion protein, three-dimensional (3D) models were obtained from AlphaFold DB7 (Varadi et al., 2024). The Shootin1 structure was further analyzed using QSalign, a homomer prediction tool available through the 3D-Beacons Network (Dey et al., 2018; Varadi et al., 2022). All protein structures were visualized and customized in PyMOL 8.
2.2. Recombinant DNA constructs
In this study, pEGFP-C1 and pCAGIG were used as host vectors, as previously described elsewhere (Zhang et al., 2019; Ergin and Zheng, 2020). All constructs were fully sequenced prior to transfection to confirm the absence of mutations introduced during molecular cloning. Plasmids were propagated in Escherichia coli DH5α cells and prepared using Qiagen miniprep kits. Recombinant plasmids expressing EGFP-fused SHTN1 and coiled–coil domain (CCD) deletion variants were generated by cloning the corresponding coding sequences into the XhoI and TspMI (New England Biolabs) sites of the pEGFP-C1 mammalian expression vector. For purification of the target proteins, pCAGIG-based constructs encoding FLAG-tagged Shootin1 were generated by polymerase chain reaction (PCR) amplification of the insert, followed by cloning into the pCAGIG vector linearized with XhoI and NotI (New England Biolabs). Oligonucleotide primer sequences used for cloning into pEGFP-C1 and pCAGIG have been reported previously [Ergin and Zheng, 2020].
2.3. Cell culture and transient expression
Neuro-2a cells [Mus musculus brain neuroblastoma; American Type Culture Collection (ATCC) #CCL-131] were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with high glucose, 10% fetal bovine serum (Thermo Fisher Scientific), and two mM GlutaMAX (Thermo Fisher Scientific), following the manufacturer’s instructions. Cells were transfected with expression plasmids, followed by coimmunoprecipitation (co-IP) or protein purification assays. Briefly, Neuro-2a cells were plated at a density of 1 × 10^6^ cells per 10-cm dish and transfected with the indicated plasmids using GeneTran-III (Biomiga), according to the manufacturer’s protocol. Forty-eight hours posttransfection, cells were harvested for downstream applications.
2.4. Co-IP and immunoblotting
To assess interactions between FLAG- and EGFP-tagged Shootin1 variants via co-IP, Neuro-2a cells transiently expressing either EGFP or FLAG-fused full-length and truncated constructs were lysed in 1 mL of lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.5% Triton X-100, protease/phosphatase inhibitor cocktails (Roche), one mM PMSF, and 100 U/mL Turbonuclease. Cell lysates were incubated with 12 μL GFP-Trap magnetic beads (Chromotek) for one h at 4 °C with gentle rotation. Beads were subsequently washed three times with 1 mL of stringent wash buffer containing 50 mM Tris-HCl (pH 7.4), 300 mM NaCl, 0.5% Triton X-100, and one mM PMSF. Bead–bound proteins were eluted by boiling in 15 μL of SDS sample buffer, and 10 μL of each sample was loaded for sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and immunoblotting with target antibodies. An additional five μL was used to probe for GFP as an expression control. Total protein lysates and immunoprecipitated fractions were analyzed by immunoblotting. Proteins were resolved on polyacrylamide gels, transferred to Immobilon-FL polyvinylidene fluoride (PVDF) membranes, and probed with primary antibodies: chicken polyclonal anti-GFP (1:2500, GFP-1020, AvesLab) and mouse monoclonal anti-FLAG M2 (1:2500, F3165, Sigma-Aldrich). Detection was performed using Alexa Fluor-conjugated secondary antibodies (Thermo Fisher Scientific; 1:2000), and blots were visualized using the GE Typhoon FLA 9000 imaging system.
2.5. Protein expression and purification
The pCAGIG-FLAG-SHTN1 construct was individually transfected into Neuro-2a cells cultured in 15-cm dishes. Forty-eight hours posttransfection, cells overexpressing FLAG-tagged SHTN1 were washed with ice-cold phosphate-buffered saline (PBS), scraped, and lysed in five mL of lysis buffer containing 50 mM Tris-HCl (pH 7.4), 500 mM NaCl, one mM ethylenediaminetetraacetic acid (EDTA), 1% Triton X-100, protease/phosphatase inhibitor cocktails (Roche), one mM phenylmethylsulfonyl fluoride (PMSF), and 100 U/mL Turbonuclease (Sigma-Aldrich). For affinity purification, 200 μL of anti-FLAG M2 magnetic bead slurry (Sigma-Aldrich), preequilibrated in lysis buffer, was added to the cleared lysate and incubated for 3 hours at 4 °C with gentle rotation. Beads were then collected using a magnetic rack and washed three times with 2 mL of lysis buffer. Bound proteins were eluted twice at room temperature for 15 min each using 1 mL of elution buffer containing 10 mM Tris-HCl (pH 7.4), 100 mM NaCl, and 200 μg/mL 3 × FLAG peptide (Apex Bio). Eluted fractions were concentrated and buffer-exchanged into Tris-buffered saline (TBS) using Amicon Ultra centrifugal filters (MWCO 30 kDa; Millipore). Purified FLAG-SHTN1 protein and BSA standards were analyzed by PAGE followed by Coomassie Brilliant Blue R-250 staining (Teknova) to assess purity and estimate the molar concentration of the target protein.
2.6. Nondenaturing native PAGE
The gel was prepared using 8% acrylamide/bis-acrylamide (30%/0.8% w/v), 0.375 M Tris-HCl, 10% ammonium persulfate, and 0.1% N,N,N′,N′-Tetramethylethylenediamine (TEMED). Protein samples were brought to 31.2 mM Tris-HCl (pH 8.8), 12.5% (v/v) glycerol, and 0.5% Bromophenol Blue, and were not heat-denatured prior to loading. The running buffer consisted of 25 mM Tris and 192 mM glycine, adjusted to pH 7.5. No SDS or reducing agents were used in any of the native PAGE buffers. Electrophoresis was performed at 200 V for four h on ice. Recombinant α-actinin, a known dimer-forming protein, was used as a positive control and processed identically to the purified FLAG-SHTN1. For Coomassie Brilliant Blue R-250 staining, gels were soaked and gently shaken in ready-to-use stain solution for 30 min, followed by destaining in 20% methanol and 7.5% acetic acid in deionized water, with repeated rinsing until the background was well cleared.
2.7. Structural modeling of the FGFR2::SHTN1 fusion complex
To construct the FGFR2::SHTN1 fusion complex model, we first predicted the dimeric structures of FGFR2 and Shootin1 independently using AlphaFold-Multimer (Evans et al., 2021). FGFR2 dimerization was modeled based on its full intracellular and transmembrane domains, whereas the Shootin1 dimer model focused on its coiled–coil domains, which mediate oligomerization. Following separate multimer predictions, the best-ranked FGFR2 and Shootin1 dimers were subjected to protein–protein docking using HADDOCK 2.4 (Honorato et al., 2024). Among the generated docking solutions, the model with the lowest HADDOCK score and highest cluster occupancy was selected for further analysis. To mimic a realistic membrane environment, the selected FGFR2::SHTN1 complex was embedded into a lipid bilayer [1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC)] using the CHARMM-GUI Membrane Builder (Wu et al., 2014). The orientation of the FGFR2 transmembrane helices was manually refined to match experimental topology. Finally, structural visualization and figure preparation were performed in UCSF ChimeraX (Meng et al., 2023), where the distinct domains of the fusion complex were colored for clarity.
2.8. Statistical analysis
Statistical analysis of co-IP experiments performed in three biological replicates was conducted using unpaired Student’s t-tests in GraphPad Prism 10. Data are presented as mean ± SEM. P-values less than 0.05 were considered statistically significant.
Results
3.1. Shootin1 is involved in diverse oncogenic modalities
Our pan-cancer analysis reveals broad dysregulation of SHTN1, with distinct alteration patterns across tumor types. We retrieved and visualized SHTN1 genomic alteration frequencies from The Cancer Genome Atlas (TCGA) Pan-Cancer Atlas studies using cBioPortal. From 10,967 patient samples spanning 32 cancer types in the Atlas, our query identified SHTN1 alterations in 154 individuals (Figure 1). Despite their presence across numerous cancers, it remains unclear whether these SHTN1 alterations represent drivers of tumorigenesis or are merely passenger events; their specific molecular correlates thus require elucidation. However, numerous studies have well established that fundamental cancer hallmarks such as cell migration, invasion, chromatin instability, and remodeling involve aberrant cytoskeleton and actin dynamics (Caridi et al., 2019; Izdebska et al., 2020; Datta et al., 2021). Given Shootin1’s roles in regulating cytoskeletal organization and actin dynamics during physiological processes, it is conceivable that SHTN1 mutations and copy number variations could disrupt these fundamental processes in certain tissues or specific microenvironments, thereby contributing to the malignant phenotype.
Shootin1’s diverse alterations indicate an intertwined, multifaceted involvement in cancer, ranging from mere presence to mechanistic collaboration. A detailed analysis of SHTN1 alterations in the cBioPortal and COSMIC databases enabled us to curate a table highlighting specific SHTN1 gene fusions, including those with reported oncogenic activity (Table). The data reveal a variety of SHTN1 fusions; however, the intrachromosomal FGFR2::SHTN1 and interchromosomal ROS1::SHTN1 fusions stand out for their well-annotated and experimentally verified roles as drivers of oncogenesis (Ross et al., 2014; Wiesner et al., 2014). Notably, both FGFR2 and ROS1 belong to the family of Receptor Tyrosine Kinases (RTKs), critical regulators of intracellular signaling pathways involved in cell growth and differentiation. Aberrant activation of such RTKs, often through gene fusions, is a well-established mechanism in oncogenesis, typically leading to constitutive kinase activity that drives uncontrolled cell proliferation. In both fusion types, SHTN1 acts as the tail gene (the 3’ partner in the fusion), resulting in an in-frame final construct. This in-frame designation indicates that the fusion retains an open reading frame within the chimeric protein, allowing the fused gene products to be translated without premature termination. The fact that SHTN1 is the tail gene suggests that the kinase domains of ROS1 and FGFR2 are potentially driving the oncogenic activity, whereas Shootin1 likely promotes the dimerization of these kinase domains.
While both ROS1::SHTN1 and FGFR2::SHTN1 fusions represent intriguing oncogenic events involving Shootin1, we will primarily focus on the FGFR2::SHTN1 fusion in this study. This is largely due to extensive literature confirming this fusion as potent and often targetable in malignancies, which also provides a rich context for mechanistic understanding. Thus, FGFR2::SHTN1 is a promising, biologically plausible candidate for developing a detailed model of Shootin1’s role in oncogenesis.
3.2. The interplay of Shootin1 and FGFR2 in cancer
FGFR2, as a pivotal RTK, normally plays critical roles in regulating cell proliferation, differentiation, and survival through tightly controlled ligand-dependent signaling. Gene fusions involving FGFR2 are well-established oncogenic drivers across numerous cancer types. These fusions typically arise from chromosomal rearrangements that juxtapose the intact kinase domain of FGFR2 with a dimerization or oligomerization domain from a partner gene, which eliminates the need for ligand binding, resulting in constitutive activation of FGFR2’s kinase activity and subsequent aberrant signaling through downstream pathways like MAPK/ERK and PI3K/Akt (Gallo et al., 2015). Such sustained activation drives uncontrolled cell growth and migration, thereby conferring significant oncogenic potential to FGFR2 fusions, which have led to their recognition as clinical targets (Parker et al., 2014; Borad et al., 2015; De Luca et al., 2020).
In this regard, CCA exhibits particularly high FGFR fusion prevalence and therapeutic potential, and importantly, FGFR2 is emerging as a significant fusion partner, along with its associate protein Shootin1 (Krook et al., 2020; Li et al., 2020; Deng et al., 2023).
CCA is a highly lethal malignancy that arises predominantly from the epithelial cells lining the bile ducts, known as cholangiocytes (Brindley et al., 2021). Given the critical role of cholangiocytes in CCA pathogenesis and the experimentally validated oncogenicity of the FGFR2::SHTN1 fusion in CCA, we investigated baseline expression of both genes in healthy human cholangiocytes using publicly available single-cell RNA sequencing data from deceased donor liver cells (MacParland et al., 2018; Speir et al., 2021). Our analysis of a t-SNE plot showing various liver cell identities revealed distinct expression patterns for FGFR2 and SHTN1 within the cholangiocyte population (Figure 2A). While FGFR2 showed robust and widespread expression across healthy cholangiocytes (Figure 2B), SHTN1 expression was notably absent within these same cells (Figure 2C). This differential expression suggests that the FGFR2::SHTN1 fusion observed in CCA indicates the presence of a de novo chimeric protein in cells that normally lack the Shootin1 component. Thus, we anticipate that this novel chimeric transcript is expressed under the control of the upstream FGFR2 promoter, given that FGFR2 serves as the 5′ fusion partner. Yet, through single-cell RNA sequencing of patient samples, further validation would fully confirm this anticipated cellular context. In this regard, we would expect single-cell data sampled from CCA biopsies to show Shootin1 as detectable, whereas FGFR2 may appear absent. This is because most single-cell RNA-seq platforms rely on polyadenylic acid capture and sequence only a short stretch from the 3′ end. Since FGFR2 is located further upstream in the fusion transcript, it may lie outside the sequencing read window and therefore go undetected. At the same time, SHTN1, positioned at the immediate 3′ end, would be captured and sequenced.
The FGFR2 and SHTN1 genes are in proximity on human chromosome 10, with FGFR2 upstream of SHTN1. The precise genetic architecture of the FGFR2::SHTN1 fusion identified in human cancers is schematically represented in Figure 3A. With the critical in-frame breakpoints, the fusion results from the joining of FGFR2 exons 1–17 to the C-terminal portion of SHTN1, encompassing its exons 7–17, thereby ensuring a continuous coding sequence (Figure 3B). Crucially, this chimeric construct retains the entire intracellular tyrosine kinase domain, a fundamental structural arrangement that positions the potent FGFR2 kinase for ligand-independent activation and oncogenic activity.
The constitutive activation conferred by the FGFR2 kinase, thus the unique oncogenic potential of FGFR2::SHTN1, is likely modulated by the incorporated C-terminal domains of Shootin1. The fusion protein specifically includes Shootin1’s coiled–coil domains (CCD-II and CCD-III) (Figure 3C). The presence of the coiled–coil domain suggests a mechanism for ligand-independent dimerization driving the constitutive activity of the fused FGFR2 kinase. This intricate molecular architecture suggests that Shootin1 possesses an inherent capacity to oligomerize via its coiled–coil domains, enabling constitutive and aberrant signaling by the oncogenic fusion protein within tumor cells.
3.3. Shootin1 oligomerizes through its coiled–coil domains
To investigate the potential role of Shootin1’s coiled–coil domains in the dimerization of the FGFR2::SHTN1 fusion, we first characterized the CCD profile of native Shootin1. In silico prediction using Marcoil (Simm et al., 2014) identified three distinct regions within the Shootin1 sequence exhibiting high coiled–coil probability scores, consistently above 90% (Figure 4A). This prediction was further supported by AlphaFold modeling, which revealed a three-dimensional structure of Shootin1 consistent with the presence of three CCDs (Figure 4B). To investigate whether Shootin1 can form oligomers, we first purified FLAG-tagged Shootin1 protein from mammalian cells. Coomassie staining of the purified samples confirmed the absence of copurifying proteins, indicating high purity of the FLAG-SHTN1 preparation (Figure 4C). We then subjected the samples to native-PAGE under nonreducing conditions. This analysis revealed that FLAG-Shootin1 migrates as a higher-order complex, consistent with an oligomeric state. As a positive control, the well-characterized dimeric protein α-Actinin exhibited a similar migration pattern, supporting the conclusion that Shootin1 oligomerizes under native conditions (Figure 4D). These findings collectively establish that Shootin1 contains three consecutive CCDs and possesses the intrinsic ability to form oligomers.
Further investigations were conducted to pinpoint the specific coiled–coil domain responsible for Shootin1’s oligomerization. We coexpressed EGFP- and FLAG-tagged Shootin1 in Neuro-2a cells and tested whether the two protein variants interacted by co-IP. Full-length EGFP-Shootin1 proteins were immunoprecipitated from cell lysates, and immunoblotting was performed to probe FLAG-tagged Shootin1 proteins. This experiment showed that the two protein variants interacted, indicating that the full-length Shootin1 molecules could form oligomers. To test whether CCD regions mediate the oligomerization, we replaced EGFP-SHTN1 with EGFP-tagged CCD deletion mutants in the co-IP experiment. Intermolecular interaction was preserved in the absence of CCD-I, but deletion of CCD-I-II (or CCD-I-II-III) completely abolished the interaction (Figure 5A–C). Since CCD-I alone is not necessary for Shootin1 oligomerization, the CCD-II region likely plays an essential role in proper oligomerization. This experimental validation was supported by in silico analyses of CCD-II. Marcoil prediction of the heptad repeat regions (Delorenzi and Speed, 2002; Simm et al., 2014) within CCD-II showed a pattern that specifically highlights the high occurrence of isoleucine or leucine residues at the ‘a and ‘d positions, which are known to promote oligomer formation (Figure 5D). Furthermore, computational modeling using CCBuilder (Wood and Woolfson, 2018) and QSalign (Dey et al., 2018; Varadi et al., 2024) predicted a preferred dimerization state for the putative CCD-II region, consistent with our experimental observations (Figure 5D and 5E). Given that the FGFR2::SHTN1 fusion construct incorporates the Shootin1 CCD-II region, these results strongly suggest that CCD-II is the primary motif driving the ligand-independent dimerization and subsequent constitutive activation of the FGFR2 kinase domain within the oncogenic FGFR2::SHTN1 chimeric protein.
3.4. Proposed model for FGFR2::SHTN1 fusion-driven oncogenic activity
The detailed characterization of the FGFR2::SHTN1 fusion’s genetic and protein architecture, combined with our findings on Shootin1’s intrinsic oligomerization capabilities, allowed us to propose a comprehensive model for its constitutive activation in cancer (Figure 6A). In its physiological state (Figure 6B, left panel), FGFR2 activation is tightly regulated and ligand-dependent. The binding of specific FGF ligands induces receptor dimerization, which in turn activates the intracellular tyrosine kinase domains. This initiates a cascade of autophosphorylation events that propagate through the downstream signaling pathways, including MAPK, STAT, AKT, and PLCγ. This controlled signaling is essential for regulating genes involved in normal cell proliferation, differentiation, and survival.
However, in the oncogenic state driven by the FGFR2::SHTN1 fusion (Figure 6B, right panel), this precise and strict regulation control is disrupted. As demonstrated by our structural and functional analyses, the FGFR2::SHTN1 fusion protein retains the FGFR2 tyrosine kinase domain intact, fused to the C-terminal portion of Shootin1, which contributes the potent coiled–coil dimerization domain CCD-II. This structural arrangement promotes ligand-independent dimerization and subsequent constitutive autophosphorylation of the FGFR2 kinase domain in the intracellular space. Such aberrant, uncontrolled activation of FGFR2 signaling then constantly drives downstream oncogenic pathways without the necessary regulatory brakes. This sustained constitutive signaling contributes significantly to malignant progression, including increased cell proliferation, enhanced invasiveness, epithelial–mesenchymal transition, and angiogenesis, which underscores the role of FGFR2::SHTN1 as a potent driver of oncogenesis.
Discussion
The identification and detailed characterization of the FGFR2::SHTN1 fusion contribute significantly to the growing understanding of oncogenic gene fusions as potent drivers in human cancer. As a family of RTKs, FGFRs are normally vital regulators of cellular proliferation, differentiation, and tissue homeostasis, but their aberrant activation frequently underpins various malignancies.
Gene fusions, in particular, represent a well-established paradigm of oncogenic drivers, and our analyses illuminate the molecular mechanism of the FGFR2::SHTN1 fusion, identifying it as a de novo oncogenic driver, particularly relevant to CCA. The precise genetic architecture, characterized by an in-frame breakpoint between upstream FGFR2 (exons 1–17) and downstream SHTN1 (exons 7–17) on human chromosome 10, highlights the critical role of the genomic architecture in facilitating this recurrent rearrangement. This specific configuration preserves the intact FGFR2 tyrosine kinase domain, with its robust expression likely driven by the native FGFR2 promoter. Our findings critically demonstrate that Shootin1 possesses intrinsic oligomerization capabilities, specifically mediated by its CCD-II domain. Our findings provide, for the first time, a direct molecular mechanism for the constitutive, ligand-independent dimerization and subsequent autophosphorylation of the fused FGFR2 kinase through the contribution of the coiled–coil domains of Shootin1 protein. The absence of SHTN1 expression in healthy cholangiocytes, in contrast to the physiological presence of FGFR2, underscores how this fusion creates an entirely novel chimeric protein with aberrant signaling potential in the cells critical for CCA initiation and progression.
While the constitutive activation of the FGFR2 kinase through Shootin1-mediated dimerization is a clear oncogenic mechanism, the unique presence of Shootin1’s actin-modulating domains (i.e., PRR, WH2, FAB) within the chimeric protein opens avenues for discussions regarding its expanded role in cancer progression. Beyond simply providing a dimerization scaffold, could the fused Shootin1 domains actively reprogram cellular architecture and dynamics? It is conceivable that the activated FGFR2 kinase might aberrantly regulate components of the actin cytoskeleton directly via its proximity to Shootin1’s domains, or conversely, that the cytoskeletal activities of the fused Shootin1 could feed back into and dysregulate FGFR2 signaling in novel ways. This could extend beyond increased proliferation to dramatically enhanced cell motility and invasiveness, through unique mechanobiological pathways. Another noteworthy possibility is that the altered cytoskeletal dynamics, driven by this fusion, may generate novel physical forces within the cell that influence drug resistance pathways.
Our previous studies demonstrated that Shootin1 is subject to partial autoinhibition mediated by its N-terminal CCD-I, which attenuates its ability to bind F-actin and may regulate its conformational dynamics (Ergin and Zheng, 2020). In addition to its role in actin regulation, CCD-I also contributes to the cytoplasmic retention of Shootin1 by masking a strong nuclear localization signal (NLS) embedded between the proline-rich region (PRR) and the WH2 domain. Deletion of the coiled–coil domains unmasks the NLS and results in robust nuclear accumulation of Shootin1, highlighting a dual regulatory role for CCD-I in both subcellular localization and cytoskeletal interactions. These regulatory mechanisms become particularly relevant in the context of the FGFR2::SHTN1 fusion identified in cancers. The fusion eliminates the N-terminal CCD-I of Shootin1, potentially releasing both autoinhibitory constraints and the NLS. As a consequence, the chimeric protein may acquire novel biochemical properties, including enhanced actin-binding activity and altered subcellular distribution. Notably, the unmasked NLS from Shootin1 could facilitate nuclear translocation of the FGFR2 moiety. A hypothesis supported by recent studies links the nuclear localization of FGFR2 to its emerging role in transcriptional regulation (Lee et al., 2019; Chen et al., 2020; Servetto et al., 2021). Therefore, the FGFR2::SHTN1 fusion may not only affect the cytoskeletal architecture through gain-of-function activity via the truncated Shootin1 protein, but also may confer nuclear functions on FGFR2, thus bridging the cytoskeletal remodeling with nuclear signaling in oncogenesis. Moreover, despite the absence of the CCD-I, the Shootin1 portion of the chimeric protein retains its full set of actin-related domains, including PRR, WH2, and the F-actin-binding domain. It is highly plausible that, while the retained CCD-II domain of FGFR2::SHTN1 facilitates ligand-independent receptor activation, the intact C-terminal domains of Shootin1 independently mediate cytoskeletal rearrangements. Together, these properties suggest that the FGFR2::SHTN1 fusion protein may act as a multifunctional oncoprotein, simultaneously driving transcriptional and cytoskeletal programs that promote tumor progression. These unique structural and functional characteristics of FGFR2::SHTN1 not only solidify its standing as a potent and targetable oncogene but also warrant further exploration of its distinct impact on cellular mechanotransduction and its potential as a highly specific diagnostic marker or a multimodal therapeutic target beyond mere kinase inhibition.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Borad MJ Gores GJ Roberts LR 2015 Fibroblast growth factor receptor two fusions as a target for treating cholangiocarcinoma Current Opinion in Gastroenterology 31 3 264 8 10.1097/MOG.0000000000000171 25763789 PMC 4750878 · doi ↗ · pubmed ↗
- 2Brindley PJ Bachini M Ilyas SI Shahid AK Loukas A 2021 Cholangiocarcinoma Nature Reviews Disease Primers 7 1 65 10.1038/s 41572-021-00300-2 PMC 924647934504109 · doi ↗ · pubmed ↗
- 3Brown LM Ekert PG Fleuren ED 2023 Biological and clinical implications of FGFR aberrations in paediatric and young adult cancers Oncogene 42 23 1875 88 10.1038/s 41388-023-02705-7 37130917 PMC 10244177 · doi ↗ · pubmed ↗
- 4Caridi CP Plessner M Grosse R Chiolo I 2019 Nuclear actin filaments in DNA repair dynamics Nature Cell Biology 21 9 1068 77 10.1038/s 41556-019-0379-1 31481797 PMC 6736642 · doi ↗ · pubmed ↗
- 5Chen MK Hsu JL Hung MC 2020 Nuclear receptor tyrosine kinase transport and functions in cancer Advances in Cancer Research 147 59 107 10.1016/bs.acr.2020.04.010 32593407 · doi ↗ · pubmed ↗
- 6Datta A Deng S Gopal V Yap KCH Halim CE 2021 Cytoskeletal dynamics in epithelial-mesenchymal transition: insights into therapeutic targets for cancer metastasis Cancers 13 8 1882 10.3390/cancers 13081882 PMC 807094533919917 · doi ↗ · pubmed ↗
- 7De Luca A Esposito Abate R Rachiglio AM Maiello MR Esposito C 2020 FGFR fusions in cancer: from diagnostic approaches to therapeutic intervention International Journal of Molecular Sciences 21 18 6856 10.3390/ijms 21186856 32962091 PMC 7555921 · doi ↗ · pubmed ↗
- 8Delorenzi M Speed T 2002 An HMM model for coiled-coil domains and a comparison with PSSM-based predictions Bioinformatics 18 4 617 25 10.1093/bioinformatics/18.4.617 12016059 · doi ↗ · pubmed ↗