Ureaplasma-driven inhibition of the epithelial Na+ transport in fetal alveolar cells: A novel mechanism of Ureaplasma-mediated preterm lung disease
Kirsten Glaser, Carl-Bernd Rieger, Elisabeth Paluszkiewicz, Ulrich H. Thome, Mandy Laube

TL;DR
This study shows how Ureaplasma infection impairs lung function in preterm infants by reducing sodium transport in fetal lung cells, potentially leading to breathing difficulties.
Contribution
The study identifies ammonia as a Ureaplasma virulence factor and shows that urease inhibitors can restore sodium transport in infected cells.
Findings
Ureaplasma infection reduces epithelial Na+ transport by 30–90% in fetal lung cells.
Ammonia from Ureaplasma, not pH shifts, mimics the infection's effects on Na+ transport.
Urease inhibitor flurofamide restores Na+ transport in infected cells.
Abstract
Respiratory tract colonization with Ureaplasma species has been repeatedly associated with the development of acute and long-term pulmonary morbidity in preterm infants. However, despite strong evidence from observational studies and animal models, apart from inflammation, underlying mechanisms of Ureaplasma-driven lung disease, such as potential functional impairments, are mainly unknown. Knowledge of Ureaplasma-lung interaction and Ureaplasma virulence factors is scarce. The present investigation is the first to examine the influence of perinatal Ureaplasma infection on critical mechanisms of alveolar fluid clearance (AFC) in immature lung cells, which drive perinatal transition from fluid-filled lungs before birth to alveolar fluid absorption, enabling lung breathing. Disruption or impairment of these mechanisms could worsen respiratory distress in preterm infants and contribute to…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
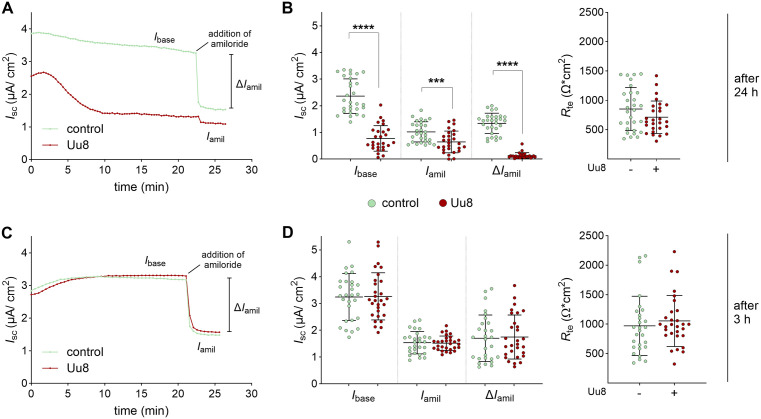 Fig 1
Fig 1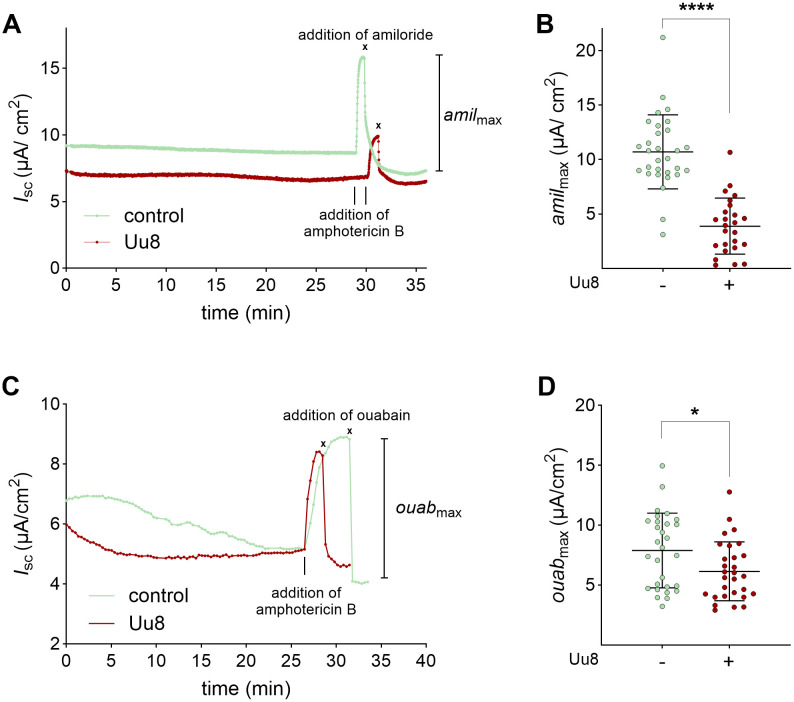 Fig 2
Fig 2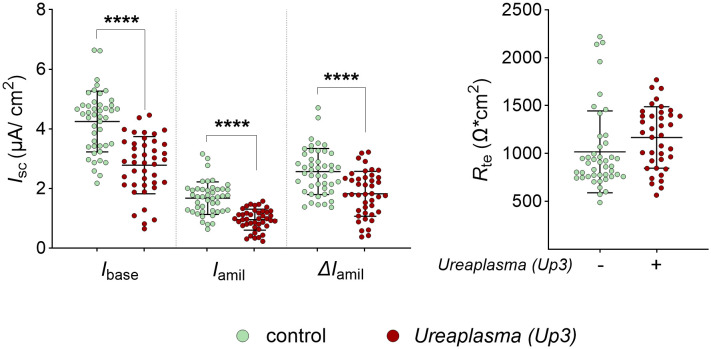 Fig 3
Fig 3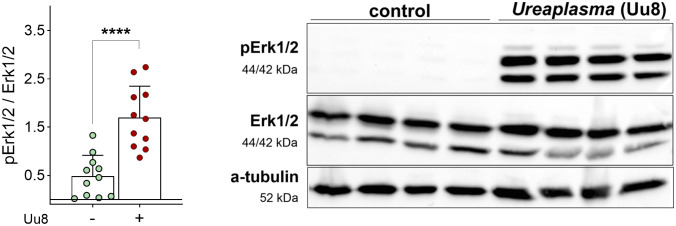 Fig 4
Fig 4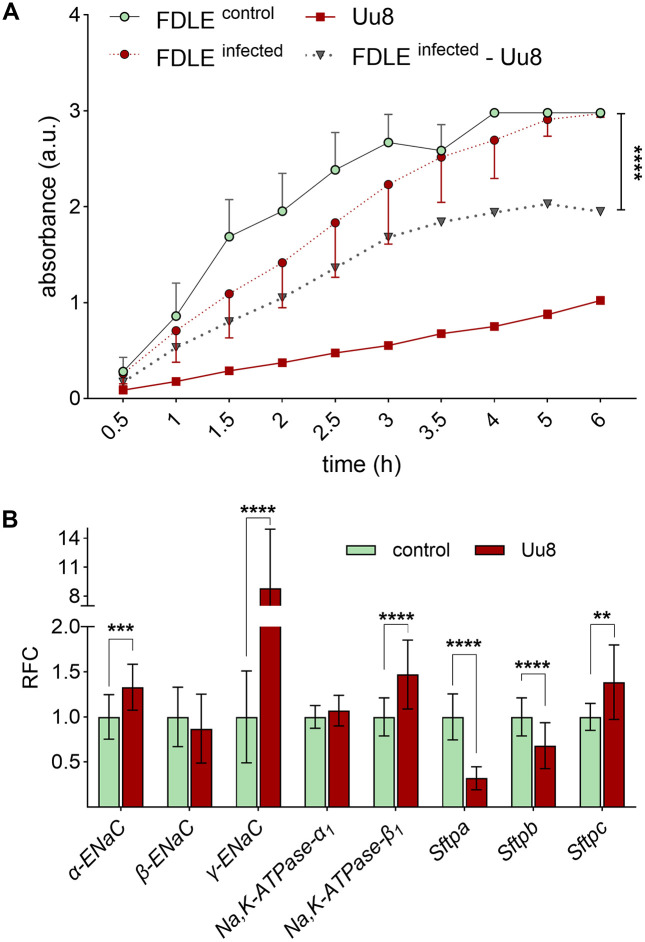 Fig 5
Fig 5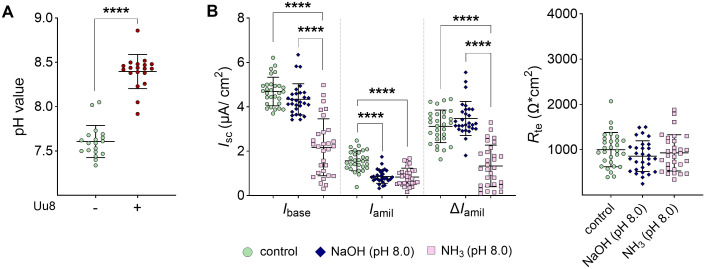 Fig 6
Fig 6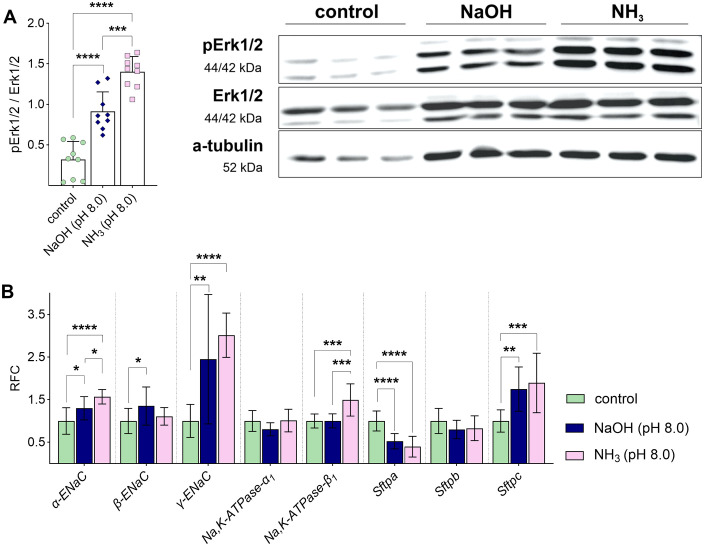 Fig 7
Fig 7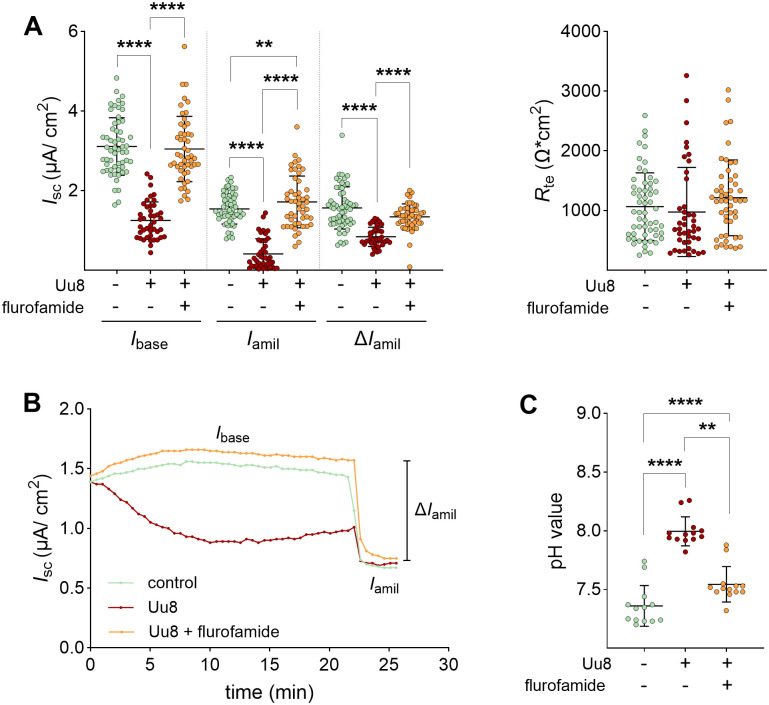 Fig 8
Fig 8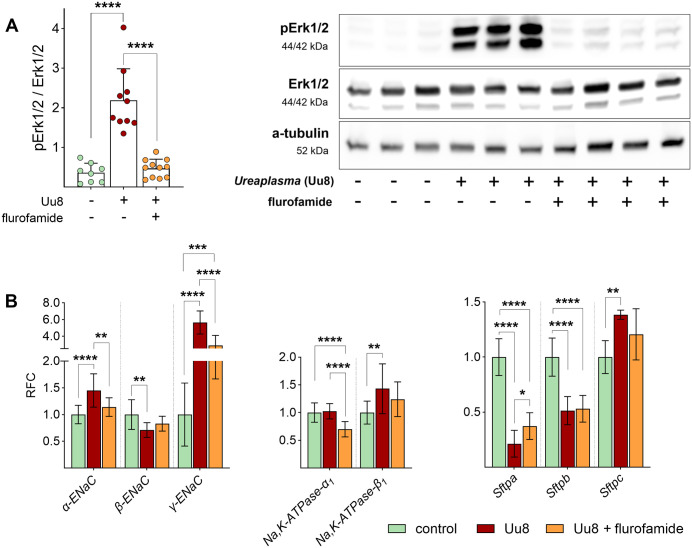 Fig 9
Fig 9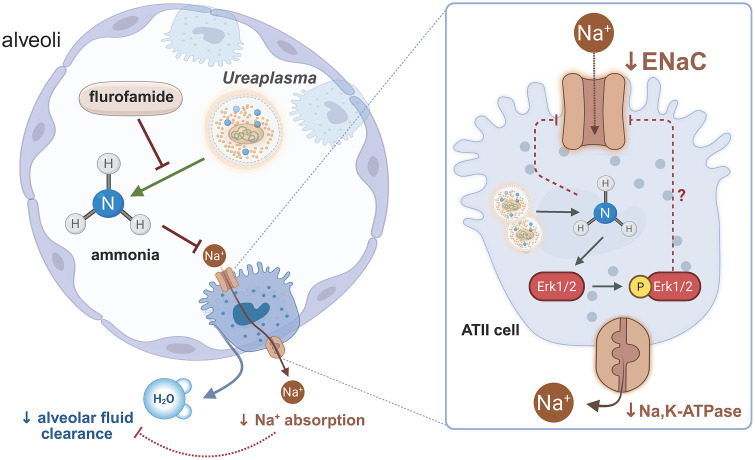 Fig 10
Fig 10- —http://dx.doi.org/10.13039/501100019173Roland Ernst Stiftung
- —http://dx.doi.org/10.13039/501100019173Roland Ernst Stiftung
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeonatal Respiratory Health Research · Preterm Birth and Chorioamnionitis · Congenital Diaphragmatic Hernia Studies
Introduction
The two Ureaplasma species, Ureaplasma (U.) parvum (serovars 1, 3, 6, 14) and U. urealyticum (serovars 2, 4, 5, 7–13), are mainly commensal bacteria of the adult urogenital tract. Yet, in preterm infants, consistent epidemiological and experimental data indicate a strong correlation between Ureaplasma respiratory tract colonization and adverse pulmonary short- and long-term outcomes, including bronchopulmonary dysplasia (BPD) [1–3]. Colonization rates increase with decreasing gestational age, leaving the most immature preterm infants at the highest risk. Noteworthy, Ureaplasma species are detected in early life respiratory tract samples of about 30% of preterm infants born below 30 weeks of gestation and up to 60% of infants born below 26 weeks [3,4]. Animal studies and in vitro data confirm inflammatory responses and altered lung development in preterm mice, sheep, rhesus macaques, and baboons exposed to intrauterine Ureaplasma infection, as well as in Ureaplasma-infected neonatal monocytes [4–6]. However, besides the indirect effects of systemic inflammation and direct apoptosis-modulating effects of Ureaplasma on pulmonary epithelial and endothelial cells [7,8], the underlying mechanisms of Ureaplasma-driven preterm acute and chronic lung disease are largely unknown. Knowledge of Ureaplasma-host interactions and Ureaplasma virulence factors is scarce. A surface-exposed family of lipoproteins termed “multiple banded antigen” has been considered the major virulence factor [9]. Hydrolysis-driven pH shift and the accompanying ammonia accumulation have long been discussed as another virulence factor potentially exerting cytotoxic effects, though remaining unexamined, so far [2,9]. Acknowledging that major morbidities, such as BPD, remain high in the most immature preterm infants in times of ever-decreasing gestational age, a better knowledge of Ureaplasma-driven preterm lung disease remains paramount to identifying addressable pathways and further developing therapeutic strategies. Notably, current antimicrobial treatment strategies may be effective in Ureaplasma eradication in preterm infants but might not prevent Ureaplasma-driven adverse neonatal outcomes.
Serovars of both Ureaplasma species have been identified in clinical samples from pregnant women and preterm infants. An association between specific species or serovars and adverse pregnancy or neonatal outcomes is not definitively established [9–12]. While some studies reported on a predominance of U. parvum [11,13,14], others have not reported any predominance of particular species or serovars [12,15]. In this study, representatives from both species were examined to evaluate potential differences in virulence.
One key mechanism driving the neonate’s adaptation to air breathing is the early postnatal alveolar fluid clearance (AFC). While intrapulmonary fluid is essential for fetal lung development, perinatally, this intrapulmonary fluid must be removed efficiently and quickly to fully establish postnatal lung function. This perinatal lung transition is impaired in preterm infants due to structural and functional lung immaturity [16]. The subsequent respiratory distress syndrome (RDS) is a leading cause of preterm morbidity and mortality [17]. Besides surfactant deficiency, an immature epithelial Na^+^ channel (ENaC) expression has been acknowledged as a critical mechanism [16]. ENaC in the apical membrane compartment of alveolar type II (ATII) cells and the basolateral Na,K-ATPase promote the epithelial Na^+^ transport [18], which osmotically drives fluid absorption from the alveolar lumen into the interstitium and circulation. Decreased AFC in preterm infants [19] is likely due to a lower ENaC expression [20]. Knockout of the α-ENaC subunit led to RDS and respiratory failure in newborn mice [21]. In ATII cells, ENaC comprises three homologous subunits, α-, β-, and γ-ENaC [18,22], while the Na,K-ATPase contains α_1_- and β_1_-subunits [22]. Perinatal impairment of ENaC function and critical AFC mechanisms in Ureaplasma-colonized alveoli could worsen respiratory distress in preterm infants and contribute to acute lung injury. The present investigation is the first to examine the potential impact of perinatal Ureaplasma infection on critical mechanisms of perinatal lung transition in preterm infants, comprising epithelial Na^+^ transport, barrier integrity, metabolic activity, gene expression, and kinase signaling. We hypothesized that perinatal alveolar Ureaplasma colonization might interfere with early-life lung function and contribute to acute lung injury in colonized preterm infants by mechanisms other than inflammation.
Results
Ureaplasma infection inhibited Na+ transport in fetal alveolar cells
A 24-h infection of FDLE cells with U. urealyticum serovar 8 (Uu8) reduced the epithelial Na^+^ transport by 70–90%. Ussing chamber analyses demonstrated that Na^+^ transport (basal ISC, Ibase) was significantly decreased by Uu8 infection compared with uninfected cells (Fig 1B). ENaC activity (amiloride-sensitive ∆ISC, ΔIamil) was also significantly reduced by Uu8 infection by more than 90%. In contrast, the epithelial barrier function, assessed as transepithelial resistance (Rte), was unaffected. Furthermore, the amiloride-insensitive ISC (Iamil), most likely Cl^-^ transport, was significantly reduced by Uu8 (Fig 1B). In contrast, 3 h incubation did not affect Na^+^ transport (Fig 1D).
*Ureaplasma infection strongly reduced epithelial Na+ transport in FDLE cells at 24 h incubation.FDLE cells were infected with serovar 8 of U. urealyticum (Uu8) for 24 h (A, B) or 3 h (C, D) before analyses. (A) Representative Ussing chamber current tracing of Uu8-infected FDLE cells versus controls after 24 h. (B) Uu8 infection significantly decreased Na+ transport and ENaC activity (Ibase, Iamil, and ΔIamil). The barrier integrity (Rte) of infected FDLE cells (n = 29) was not significantly altered compared to uninfected controls (n = 27). (C) Representative current tracing of Uu8-infected FDLE cells versus controls after 3 h. (D) Infection of FDLE cells with Uu8 for 3 h did not affect Ibase, Iamil, ΔIamil or Rte (n = 29) compared to control cells (n = 28). Statistical analysis: ***p < 0.001, ***p < 0.0001 by unpaired t-test.
To consider the capacities of ENaC and the Na,K-ATPase separately, the opposite membrane was permeabilized. The maximum amiloride-sensitive apical membrane permeability (amilmax) was significantly decreased by Uu8 to approximately 35% (Fig 2B). In addition, the maximum ouabain-sensitive Na,K-ATPase activity (ouabmax) was significantly reduced upon Uu8 infection, although not to the same extent (Fig 2D). In summary, Uu8 diminished the apical ENaC activity by more than 60% and the basolateral Na,K-ATPase activity by approximately 20% in FDLE cells upon 24 h infection.
*Ureaplasma infection reduced isolated maximal ENaC and Na,K-ATPase activities.FDLE cells were infected with Uu8 for 24 h. The maximal activity of each Na+ transporter was determined separately by permeabilizing the opposite membrane. (A) Representative current tracings of basolaterally permeabilized Uu8-infected or control cells, measured with a 145:5 apical-to-basolateral Na+ gradient. (B) Uu8 infection significantly reduced the maximum apical Na+ permeability mediated by ENaC (amilmax) (n = 30) compared to controls (n = 25). (C) Representative current tracings of apically permeabilized Uu8-infected or control cells. (D) Maximum Na,K-ATPase activity (ouabmax) was significantly reduced by Uu8 infection (n = 29) compared to uninfected FDLE cells (n = 30). Statistical analysis: *p < 0.05, ***p < 0.0001 by unpaired t-test.
To determine if the effects observed for Uu8 were serovar-specific, the electrophysiological measurements were repeated with serovar 3 of U. parvum (Up3). Under the same culture conditions, Up3 showed less growth in the same period, resulting in lower pre-infection titers than those obtained for Uu8. However, as seen before, Up3 significantly decreased epithelial Na^+^ transport and ENaC activity in infected FDLE cells compared to uninfected controls (Fig 3). Compared to Uu8, the reduction of Na^+^ transport by Up3 was less pronounced, attenuating ENaC activity by approximately 30%.
*Parallel infection with a Ureaplasma parvum serovar 3 reproduced the observed effects on the epithelial Na+ transport.Infection with serovar 3 of U. parvum (Up3) also reduced epithelial Na+ transport in FDLE cells. Cells were infected with Up3 for 24 h before analyses. In line with Uu8 infection, Up3 infection significantly decreased Na+ transport and ENaC activity (Ibase, Iamil, and ΔIamil), while Rte was unaffected (Up3: n = 45; control: n = 42). Statistical analysis: ***p < 0.0001 by unpaired t-test.
Ureaplasma infection induced phosphorylation of Erk1/2
To investigate whether signaling pathways were disturbed by Ureaplasma infection, the phosphorylation of p44/42 MAPK (Erk1/2) was analyzed by Western blot. Erk1/2 is a well-known inhibitor of ENaC function. As given in Fig 4, Uu8 infection strongly increased phosphorylation of Erk1/2 in FDLE cells by more than 3-fold (Figs 4 and S1), possibly contributing to the diminished ENaC activity.
*Ureaplasma infection significantly activated Erk1/2 signaling in FDLE cells.Erk1/2 phosphorylation was significantly induced upon Uu8 infection (n = 11) compared to controls (n = 11). Representative Western blots give pErk1/2, total Erk1/2 and α-tubulin as loading control. Statistical analysis: ***p < 0.0001 by unpaired t-test.
Ureaplasma infection impaired metabolic activity and modulated gene expression in FDLE cells
The metabolic activity was determined by the Water-Soluble Tetrazolium 1 (WST-1) assay in Uu8-infected FDLE cells, uninfected controls, and viable Uu8 isolates. Comparing Uu8-infected FDLE cells with uninfected controls, no significant difference was observed (Fig 5A). However, viable Ureaplasma themselves reduced tetrazolium salts even in the absence of FDLE cells. We therefore subtracted the results obtained for Uu8 from those obtained for Uu8-infected FDLE cells and found a significantly impaired metabolic activity in FDLE cells upon infection (Fig 5A).
*Ureaplasma infection significantly affected the metabolic activity and mRNA expression of FDLE cells.(A) Metabolic activity was measured every 30 min to 1 h in Uu8-infected FDLE cells, uninfected controls and viable Uu8 isolates. The dotted line represents the metabolic activity of Uu8-infected FDLE cells subtracted by the metabolic activity exhibited by Uu8 themselves. Based on this calculation, the metabolic activity was significantly lower in Uu8-infected FDLE cells than in uninfected controls (n = 8-12 per group and time point). (B) Uu8 infection significantly increased mRNA expression of α- and γ-ENaC, Na,K-ATPase-β1, and Sftpc. In contrast, Uu8 infection downregulated Sftpa and Sftpb mRNA expression (n = 12-18). Relative fold change (RFC). Statistical analysis: ****p < 0.0001 by repeated measures two-way ANOVA with Geisser-Greenhouse correction (A) or **p < 0.01, ***p < 0.001, ***p < 0.0001 by unpaired t-test (B).
Surprisingly, Uu8 infection significantly increased mRNA expression of the subunits α-ENaC by approximately 30% and γ-ENaC by almost 9-fold, while β-ENaC mRNA expression was unaffected (Fig 5B). Moreover, Uu8 infection significantly increased mRNA expression of the Na,K-ATPase-β1-subunit by nearly 50%, while the α1-subunit level was not altered. In contrast, the surfactant protein A (Sftpa)‘s mRNA expression decreased by approximately 70% and the expression of Sftpb by around 30% (Fig 5B). The opposite effect was observed for the Sftpc mRNA, increasing by almost 40%. Taken together, the Uu8 infection of FDLE cells increased the mRNA expression of several Na^+^ transporter subunits, in contrast to the observed functional impairment of the epithelial Na^+^ transport.
Ammonia exposure and not pH shift mimicked Ureaplasma-driven effects
A 24-h infection of FDLE cells with Ureaplasma caused a relevant pH shift (Fig 6A) due to Ureaplasma-driven hydrolysis of urea to ammonia (NH_3_) and the subsequent alkalization of the culture medium. We tested whether the pH shift or NH_3_ was responsible for the observed effects in Ureaplasma-infected FDLE cells. The pH shift to 8.0 using NaOH did not affect Na^+^ transport or ENaC activity, whereas adding NH_3_ to FDLE cells strongly reduced the Na^+^ transport (Fig 6B). Taken together, Ureaplasma-induced NH_3_ and not the accompanying pH shift inhibited Na^+^ transport and ENaC activity.
*Ureaplasma-driven NH3 mimicked the effects of acute infection on the epithelial Na+ transport.Ureaplasma infection shifted the pH in infected FDLE cells due to NH3 production. This NH3 accumulation mimicked the impact of Ureaplasma infection to a large extent. (A) Uu8 significantly increased the pH of the FDLE cell medium after 24 h (n = 19 each). (B) The pH of the FDLE cell medium was shifted to 8.0 by adding either NaOH or NH3 and maintained for 24 h. Exposure of FDLE cells to NH3 (n = 30) significantly decreased Na+ transport and ENaC activity (Ibase, Iamil, and ΔIamil) compared to control cells (n = 29), while NaOH-driven pH shift (n = 29) only affected Iamil. Rte was neither affected by NaOH-driven pH shift nor NH3. Statistical analysis: ***p < 0.0001 by unpaired t-test (A) or one-way ANOVA with Tukey’s multiple comparison test (B).
Moreover, phosphorylation of Erk1/2 was increased by NaOH-driven pH shift and NH_3_, but the effects of NH_3_ exposure were significantly more pronounced (Figs 7A and S2). In contrast, the Ureaplasma-driven increase of α-ENaC, γ-ENaC, and Sftpc mRNA expression was mimicked by both the NaOH-driven pH shift and the exposure to NH_3_ (Fig 7B). In addition, the NaOH-driven pH shift and NH_3_ exposure both downregulated Sftpa mRNA expression (Fig 7B).
*NaOH- and NH3-driven pH shift significantly affected Erk1/2 signaling and modulated mRNA expression.FDLE cells were incubated with media containing NaOH or NH3 at pH 8.0 for 24 h before analyses. (A) NaOH- and NH3-driven pH shift to 8.0 significantly increased Erk1/2 phosphorylation (n = 9 each). Representative Western blots of pErk1/2, total Erk1/2 and α-tubulin are given. (B) NaOH- and NH3-driven pH shift to 8.0 significantly increased mRNA expression of α- and γ-ENaC, Na,K-ATPase-β1, and Sftpc. In contrast, NaOH and NH3 downregulated Sftpa mRNA expression (n = 11-12). Statistical analysis: *p < 0.05, **p < 0.01, ***p < 0.001, ***p < 0.0001 by one-way ANOVA with Tukey’s multiple comparison test.
Flurofamide prevents Ureaplasma-driven impairment of Na+ transport and Erk1/2 phosphorylation
Flurofamide is a known inhibitor of bacterial urease. According to the findings above, we hypothesized a role for the Ureaplasma-specific urease as a key virulence factor in our experimental setting that might be affected by flurofamide. Thus, FDLE cells were co-incubated with Uu8 and the urease inhibitor to test its ability to restore the Na^+^ transport. Uu8-driven inhibition of the Na^+^ transport was entirely prevented by flurofamide (Fig 8A). Notably, flurofamide ameliorated ENaC activity in Uu8-infected cells to control levels. In addition, flurofamide attenuated the pH shift in Uu8-infected cells (Fig 8C).
*The presence of flurofamide fully restored the Ureaplasma-driven reduction of epithelial Na+ transport.FDLE cells were infected with Uu8 in the presence or absence of flurofamide (10 µM) for 24 h. (A) Uu8 infection (n = 42) significantly decreased Na+ transport and ENaC activity (Ibase, Iamil, and ΔIamil) compared to uninfected controls (n = 57). These findings were entirely restored by co-incubating FDLE cells with flurofamide (n = 49). Rte was neither affected by Uu8 infection nor flurofamide. (B) Representative Ussing chamber current tracing of FDLE cells. (C) Flurofamide significantly downregulated media pH in Uu8-infected FDLE cells (n = 13 each). Statistical analysis: **p < 0.01, ***p < 0.0001 by one-way ANOVA with Tukey’s multiple comparison test.
Notably, flurofamide prevented the Uu8-induced phosphorylation of Erk1/2 (Figs 9A and S3), possibly contributing to the restored ENaC activity. Finally, the Uu8*-induced increase of α-ENaC and γ-ENaC mRNA expression was significantly reduced by flurofamide (Fig 9B). Sftpa mRNA expression was marginally elevated by flurofamide in Uu8-*infected FDLE cells.
*Co-incubation with the urease inhibitor flurofamide prevented Ureaplasma-induced phosphorylation of Erk1/2.FDLE cells were infected with Uu8 in the presence or absence of flurofamide (10 µM) for 24 h. (A) Addition of flurofamide prevented the Uu8-driven Erk1/2 phosphorylation. Uu8 infection significantly increased Erk1/2 phosphorylation, while elevated phosphorylation was not seen in the presence of flurofamide (n = 8 each). Representative Western blots of pErk1/2, total Erk1/2 and α-tubulin are given. (B) Flurofamide had a marginal impact on Uu8-induced changes in mRNA expression of Na+ transporters and surfactant proteins. While Uu8 infection significantly increased mRNA expression of α- and γ-ENaC, Na,K-ATPase-β1, and Sftpc, flurofamide diminished the Uu8-induced increase of α- and γ-ENaC and Sftpc. Uu8 infection downregulated mRNA expression of Sftpa and Sftpb, which was not affected by flurofamide (n = 12 each). Statistical analysis: *p < 0.05, **p < 0.01, ***p < 0.001, ***p < 0.0001 by one-way ANOVA with Tukey’s multiple comparison test.
Discussion
To our knowledge, this is the first study to examine potential direct effects of Ureaplasma species on fetal alveoli and to describe a profound impairment of the epithelial Na^+^ transport in fetal alveolar cells upon acute Ureaplasma infection. Respiratory tract colonization of preterm infants with Ureaplasma has been repeatedly associated with adverse pulmonary outcome, especially BPD [1–3]. In contrast to the adult respiratory tract, the alveoli of preterm infants – filled with urea-enriched amniotic fluid until the first hours of life – provide a unique and highly favorable environment for Ureaplasma species. Since the epithelial Na^+^ transport is essentially involved in perinatal lung transition to air breathing, we sought to determine the potential impact of a perinatal Ureaplasma infection on Na^+^ transport-associated mechanisms of AFC.
In FDLE cells, an established model of otherwise unavailable preterm alveoli, we could separate the effects on basolateral Na,K-ATPase, and apical Na^+^ channels by selective permeabilization and observed a significant inhibition of the apical ENaC activity and a less profound reduction of the Na,K-ATPase activity induced by Ureaplasma species. While this robust ENaC inhibition emerged upon 24-hour infection, no effect was observed at 3 hours, which is in accordance with in vitro studies in other models reporting epithelial injury and inflammatory signaling evident by ~24 hours [8,23], although Ureaplasma seem to adhere within a few hours [24]. Notably, diminished epithelial Na^+^ transport in preterm infants [19] has been shown to contribute to the development of RDS [16]. According to our findings, functional impairment of the Na^+^ transport in Ureaplasma-colonized infants could further aggravate the critical early-life respiratory situation and worsen the respiratory short- and long-term outcome.
Since 2002, an adjusted taxonomy discriminates the two human species U. parvum and U. urealyticum. Notably, serovars of both species are found in specimens of pregnant women and preterm infants, and controversy remains whether one of the two species or individual serovars are more frequently associated with diseases than others [9,12]. To investigate potential species-specific pathogenicity, representatives of both species were used in the present study. We confirmed that both the U. parvum and the U. urealyticum serovar profoundly affected the epithelial Na^+^ transport. However, the U. urealyticum serovar exhibited a more potent inhibition, reducing the ENaC activity by 90%, while the U. parvum serovar achieved a lesser reduction of about 30%. This less profound effect might not correlate with a generally smaller effect on fetal alveolar cell function but is most likely due to lower titers of Up3 in our experiments. However, overall, the range of Ureaplasma inocula used in this study corresponded to concentrations in vivo with a Ureaplasma load reported in the cervical fluid of pregnancies complicated by preterm labor ranging from 5.0x10^6^ - 1.5x10^8^ cn/ml and the Ureaplasma load determined in the amniotic fluid of preterm births ranging from 4.0x10^3^ – 5.2x10^7^ cn/ml [25,26].
Western blot analyses revealed Ureaplasma-induced phosphorylation of Erk1/2, a signaling molecule significantly involved in regulating ENaC activity in previous studies. Acknowledging that phosphorylation of Erk1/2 has been shown to diminish ENaC activity in renal cells [27], we speculate that Ureaplasma-driven Erk1/2 phosphorylation represents one possible mechanism contributing to the observed ENaC inhibition. This is consistent with published findings in other epithelial systems: ERK activation decreases ENaC open probability without altering surface abundance, resulting in functional inhibition [28,29]. Additionally, ERK activity can promote NEDD4L (neural precursor cell expressed, developmentally downregulated 4)-mediated ubiquitination and degradation of ENaC subunits, thereby reducing channel surface density [30,31]. Importantly, both mechanisms converge on reduced amiloride-sensitive Na⁺ transport, which we directly demonstrate.
WST-1 assays demonstrated a significantly lower metabolic activity in Ureaplasma-infected cells that might indicate augmented cytotoxicity. These findings align with the Ureaplasma-driven pro-apoptotic and apoptosis-modulating effects described in pulmonary epithelial and endothelial cells [7,8]. In our WST-1 assays, we demonstrated that Ureaplasma isolates themselves reduced tetrazolium salts to formazan even in the absence of host cells. Subsequently, the Ureaplasma-inherent metabolic activity was subtracted from the results assessed in Ureaplasma-infected FDLE cells. Our findings align with genomic sequence analysis*,* suggesting that NADH, reducing the tetrazolium salt WST-1 in the given assay, contributes to several enzyme reactions in Ureaplasma metabolic pathways [32].
It is worth noting that the reduced activity of both Na^+^ transporters in FDLE cells was not due to diminished mRNA expression. In fact, the mRNA expression of the α- and γ-ENaC subunits was increased, possibly reflecting a positive feedback mechanism. The functional relevance of each subunit might well explain the observed expression patterns of the ENaC subunits. In the fetal ovine lung, the α- and γ-ENaC subunit mRNA expression peaked during labor [33], indicating that these ENaC subunits are critical at the developmental stage of the FDLE cells. On the other hand, overexpression of β-ENaC in mice decreases mucus clearance and reduces fluid in the postnatal lung, resulting in a cystic fibrosis-like phenotype [34]. The absent response of β-ENaC mRNA expression in FDLE cells in the present study might be explained by its relevance mainly for postnatal lung function.
Notably, this study showed an increased mRNA expression of the Na,K-ATPase-β1 subunit in FDLE cells upon Ureaplasma infection. Overexpression of the β1-subunit was previously demonstrated to increase vectorial Na^+^ transport [35], with this subunit being the rate-limiting component in the assembly of the Na,K-ATPase [36]. In the present study, one may speculate that an upregulation of Na,K-ATPase-β1 mRNA expression counteracts Ureaplasma-driven diminished Na^+^ transport.
One potential virulence factor of Ureaplasma often discussed in the context of Ureaplasma-host cell interaction is the urease-mediated generation of NH_3_ and the accompanying pH shift [2,9]. Although frequently considered, these potential virulence factors have not been investigated so far. The present study is the first to describe Ureaplasma-derived NH_3_ as a Ureaplasma virulence factor in a host-cell specific context affecting the epithelial Na^+^ transport fetal alveolar cells. While a NaOH-mediated pH shift did not affect ENaC activity, exposure of FDLE cells to NH_3_ significantly inhibited the epithelial Na^+^ transport, mimicking Ureaplasma-driven effects. These findings demonstrate that the observed effects cannot be attributed to alkalinization per se, but require the presence of NH_3_, representing the biologically active species mediating the observed effects. Moreover, NH_3_ exposure also affected mRNA expression of Na^+^ transporters and surfactant proteins and induced Erk1/2 phosphorylation, as observed for the incubation of FDLE cells with viable Ureaplasma isolates*.* Notably, a previous report identified two ammonia-specific upregulated phosphorylation sites on Thr202 and Tyr204, known to be phosphorylated by MAP2K1/2, causing activation of ERK1 [37]. In this study using MCF-7 breast cancer cells, quantitative phospho-proteomics strongly suggested that ERK is activated in response to ammonia treatment. Another study supported this finding, showing increased ERK1/2 phosphorylation in response to ammonium and ammonia in MCF-7 cells [38]. Notably, urease-driven virulence has been well described in other urease-expressing pathogens, such as Proteus mirabilis [39,40], contributing to epithelial dysfunction and tissue injury, for instance, in urinary tract infections [40,41].
To further delineate the relationship between Na^+^ transport and Ureaplasma-driven NH_3_ production, we inhibited the responsible Ureaplasma enzyme urease with flurofamide, an urease inhibitor approved by the FDA (Food and Drug Administration). Following our hypothesis, flurofamide completely restored Na^+^ transport and ENaC activity in Ureaplasma-infected FDLE cells. In addition, flurofamide prevented the Ureaplasma-driven phosphorylation of Erk1/2. This aligns with studies confirming that flurofamide inhibits NH_3_ production from urea by intestinal microorganisms in vitro [42]. Notably, Ureaplasma-driven NH_3_ has been implicated in the pathogenesis of hyperammonemia syndrome in infected immunocompromised patients [43,44]. A mouse model confirmed fatal *Ureaplasma-*induced hyperammonemia in the context of immunosuppression [45] and described reduced blood ammonia levels upon flurofamide treatment [46]. In line with these data, our findings suggest that flurofamide or other urease-inhibiting agents might be promising candidates for future non-antibiotic treatment strategies in Ureaplasma-colonized preterm infants. This is especially important since no treatment is available to improve Na^+^ transport and AFC during perinatal lung transition. Moreover, current antimicrobial treatment strategies, mainly using macrolide antibiotics, have several potential side effects and may not prevent long-term adverse neonatal outcomes, according to most recent data [47].
Finally, the gene expression of surfactant proteins was modulated differently in Ureaplasma-infected FDLE cells in the present study. Ureaplasma-induced downregulation was observed for Sftpa and Sftpb mRNA expression, while Sftpc expression was increased. SP-A contributes to the innate immune system by opsonization, promoting the uptake of various microorganisms by phagocytic cells [48]. Moreover, SP-A protects the alveolar region from uncontrolled inflammatory responses by reducing proinflammatory Toll-like receptor signaling [48]. We speculate that the observed Ureaplasma-induced reduction of Sftpa expression in our study could possibly weaken alveolar defense. Notably, the present study is the very first investigating a potential impact of Ureaplasma respiratory tract infection on alveolar surfactant synthesis, and this mechanism has not been described before. Of interest, Ureaplasma-driven downregulation of Sftpa might also impair the host’s immune defense against Ureaplasma. SP-A was demonstrated to enhance ureaplasmacidal activity in murine alveolar macrophages [49]. SP-B and SP-C are essential for the surface-spreading properties of pulmonary surfactant. Still, only the lack of SP-B is incompatible with life, while SP-C is supposed to contribute to stabilization of surfactant molecule effects on the alveolar structure [48]. In the present study, Ureaplasma infection downregulated the indispensable expression of Sftpb, potentially further contributing to preterm lung disease. It is worth mentioning that the present data are in line with investigations on lipopolysaccharide (LPS)-induced lung inflammation and lung injury. The latter demonstrated that LPS-induced lung dysfunction corresponds closely to abnormal surfactant function and reduced alveolar Sftpb mRNA and SP-B protein expression [50].
In summary, this is the very first investigation to determine potential direct interactions of Ureaplasma with immature alveoli and to reveal significant functional impairments of alveolar epithelial cells upon acute Ureaplasma infection. The present study suggests a global inhibition of the epithelial Na^+^ transport in fetal alveolar cells (Fig 10), potentially due to altered Erk1/2 signaling. Ureaplasma-driven accumulation of NH_3_ was identified as a key contributing virulence factor, mimicking most *Ureaplasma-*driven effects on FDLE cell function. In line with this observation, the urease inhibitor flurofamide completely restored Na^+^ transport, ENaC activity, and Erk1/2 activity, confirming the role of the Ureaplasma-driven hydrolysis of urea as virulence factor in this context.
Summarized suggested mechanism of Ureaplasma-driven inhibition of Na+ transport, impairment of perinatal AFC and transition to air breathing.Our data demonstrate a Ureaplasma-driven inhibition of Na+ transport in fetal alveolar cells. Moreover, the Ureaplasma-driven ammonia production induces Erk1/2 phosphorylation. This might be an underlying mechanism of the observed inhibition of ENaC activity and Na+ transport in Ureaplasma-infected alveolar epithelial cells. Notably, flurofamide, an inhibitor of the Ureaplasma-specific enzyme urease, restores Na+ transport and normalizes Erk1/2 phosphorylation in Ureaplasma-infected FDLE cells. Created in BioRender. Laube, M. (2025) https://BioRender.com/x30p361.
Some limitations of this study ought to be acknowledged. We used primary fetal alveolar epithelial cells of rat origin since primary or immortalized human fetal or neonatal cells originating from the deeper respiratory tract of immature preterm infants are unavailable. The given model was the best approximation for the very first study on potential functional impairments upon preterm Ureaplasma respiratory tract infection, especially since fetal rat lungs at the saccular stage used in the present study exhibit features of immaturity that are very similar to the underdeveloped lung structures of human preterm infants. Moreover, in the present study, FDLE cells were cultured under medium-submerged conditions that resemble the fetal intrauterine environment, where the developing lung is filled with urea-rich fluid, providing an optimal niche for Ureaplasma survival and proliferation. However, the current findings need to be confirmed in other models. In particular, this study ought to promote investigations in a preterm animal model of Ureaplasma respiratory tract infection that allows assessment of respiratory function and testing of potential effects of intra-alveolar application of the urease inhibitor flurofamide in Ureaplasma-infected animals. Further research in the given in vitro setting will include the incorporation of neonatal immune cells, mimicking the presence of alveolar macrophages, and will investigate the effect of other bacteria potentially present perinatally in the preterm alveoli but not generating NH_3_ as Ureaplasma species do. The two existing Ureaplasma species were both tested in the present study. However, we acknowledge that pre-selecting specific serovars may have caused selection bias.
Finally, we would like to clarify that Western blot analysis of ENaC subunits was not included in this study since reliable detection of ENaC proteins in lung tissue or primary alveolar epithelial cells is technically challenging, especially in the fetal state, where ENaC expression is shifting as the lung transitions from fluid secretion in utero to fluid absorption at birth [51,52]. In contrast to kidney or colon epithelia, ENaC expression in the lung is relatively low and developmentally regulated, often resulting in weak and variable bands that are hardly quantitatively interpretable. This limitation has been well documented in previous studies (e.g., [29,53–55]. Therefore, we focused on functional measurements of amiloride-sensitive Na⁺ transport, which directly reflect ENaC activity at the apical membrane and are considered the gold standard for assessing ENaC-mediated Na⁺ transport.
To the best of our knowledge, this is the first study to describe a functional impairment of fetal pulmonary epithelial cells upon acute Ureaplasma infection and to reveal potential mechanisms of Ureaplasma-driven preterm lung disease. Moreover, this is the first investigation to describe Ureaplasma-mediated NH_3_ accumulation as the causative Ureaplasma virulence factor. Notably, the present findings might have a significant translational impact. In preterm infants with perinatal alveolar colonization with Ureaplasma species, Ureaplasma-mediated inhibition of epithelial Na^+^ transport could impair the essential AFC, contribute to lung fluid accumulation, and worsen the respiratory distress of preterm infants. Ultimately, this study underlines the relevance of Ureaplasma as true pathogens in preterm infants, especially the most immature ones, and sheds light on the role of urease-inhibiting drugs as potential therapeutic strategies in colonized preterm infants early in life. Urease inhibitors might constitute useful alternatives to antimicrobial strategies in the future. Further studies ought to confirm the observed mechanisms in vivo and investigate their role as addressable virulence factors in the clinical context.
Materials and methods
Ethics statement
All experimental protocols were approved by the institutional review board (IRB: Landesdirektion Leipzig, Leipzig, Germany, permit number: TVV15/22). All methods were performed in compliance with Directive 2010/63/EU.
Bacterial strains and culture conditions
U. parvum serovar 3 (Up3) and U. urealyticum serovar 8 (Uu8), both often associated with disease manifestation [15,56], were obtained from the American Tissue Culture Collection (ATCC, European distributor LGC Standards GmbH, Wesel, Germany; Up3: ATCC 27815, Uu8: ATCC 27618). Frozen stocks were prepared from the mid-logarithmic phase. For each experiment, isolates were inoculated 1:10 with in-house medium containing 82% autoclaved PPLO medium (Becton, Dickinson & Company, Franklin Lakes, NJ, USA), 11% heat-inactivated horse serum (v/v), urea (186 mM, Merck, Taufkirchen, Germany) and 1% phenol red (Merck), and adjusted to pH 6.5 [5,6]. Serial dilutions of both strains were incubated overnight. Determination of color-changing units (CCU) was performed in 96-well plates (Greiner Bio-One, Frickenhausen, Germany) by 10-fold serial dilutions in 200 µl broth [57]. Uu8 and Up3 DNA copy numbers (cn) were determined by qPCR (Table 1). We tested the effects of a 3-hour infection of FDLE cells with Ureaplasma to assess potential immediate pathogen-cell interactions and a 24-hour infection to investigate the impact on established cellular and functional responses.
Table 1: Ureaplasma primer sequences.
Isolation and stimulation of FDLE cells
Sprague-Dawley rats were bred at the Medical Research Center of the University of Leipzig. Pregnant rats were euthanized by pentobarbital injection on gestation days E20-21 (term E = 22). A fetal rat lung at E20-21 is in the saccular stage of development, which closely overlaps with the developmental stage of human preterm infants at 24–36 weeks of gestation. Each FDLE cell preparation consisted of two pregnant rats with approximately 30–40 fetal rats of both sexes. The primary rat FDLE cells, a model of preterm ATII cells, were isolated from rat fetal lungs [35]. The fetal lungs were mechanically and enzymatically dissociated, and the resulting cell suspension was subjected to differential centrifugation and selective plating to enrich for epithelial cells with a resulting epithelial purity of > 95%, as previously described [58,59]. The obtained FDLE cells were seeded onto permeable Snapwell inserts (surface area 1.1 cm^2^, Corning, Corning, NY, USA) at a density of 1x10^6^ cells per insert for electrophysiological measurements. For RNA isolation, cells were seeded onto larger inserts (ThinCert, 4.6 cm^2^ surface area, Greiner Bio-One) at a density of 2x10^6^ cells per insert. For all experiments, FDLE cells were cultured under submerged conditions, reproducing the physiological context of the perinatally fluid-filled alveoli. Standard cell culture medium consisted of MEM with 10% fetal bovine serum (FBS, Biochrom, Berlin, Germany), glutamine (2 mM, Life Technologies), and antibiotic-antimycotic (Life Technologies). Four days after isolation, cells were incubated with Uu8 and Up3 cultures corresponding to 10^6^ and 10^8^ CCU/ml viable organisms (corresponding to 1.3x10^6^ – 1.8x10^7^ cn/ml), respectively, diluted 1:5 in cell culture medium for 3 or 24 hours (h), or cultured with NaOH or NH_4_OH (referred to as NH_3_) at pH 8.0 for 24 h. Since NH_3_ exists in rapid pH-dependent equilibrium with NH_4_^+^ in buffered solutions, all exposures represent a mixed NH_3_/NH_4_^+^ system. In the present study, addition of ammonium hydroxide (≈ 13.4 M) to the cell culture medium resulted in a nominal total NH_3_/NH_4_^+^ concentration of ~6.7 mM. At pH 8.0, approximately 5–6% of this pool is present as uncharged NH_3_ (pK ≈ 9.25), corresponding to ~0.35 mM NH_3_. Sealing of the culture plates prevented the medium buffer from neutralizing the pH. The impact of varying Ureaplasma inocula was tested in preliminary dose-response experiments using dilutions of 1:5, 1:10, and 1:50 and assessing epithelial Na^+^ transport and transepithelial resistance. Based on maximum effects on Na^+^ transport, minimum impact on epithelial barrier integrity, and in vivo amnion fluid Ureaplasma concentrations, we used 1:5 dilutions of the given Uu8 and Up3 cultures throughout this study. Finally, co-incubation with flurofamide (10 µM, Merck) was used to inhibit the Ureaplasma-specific urease. All experiments were performed with FDLE cell cultures obtained from three independent preparations.
Ussing chamber analyses
Ussing chamber measurements [60] were performed following a 3- and 24 h incubation with Ureaplasma. Only cell monolayers with a Rte exceeding 300 Ω·cm^2^ were included in the analyses. Electrophysiological solutions consisted of (in mM) 145 Na^+^, 5 K^+^, 1.2 Ca^2+^, 1.2 Mg^2+^, 125 Cl^−^, 25 HCO^3−^, 3.3 H_2_PO_4_^−^ and 0.8 HPO_4_^2−^ (pH 7.4), with 10 glucose (basolateral) or 10 mannitol (apical). The solutions were continuously gassed with carbogen (5% CO_2_, 95% O_2_) during measurements. Equivalent short-circuit currents (ISC) were determined every 20 s by measuring transepithelial voltage (Vte) and Rte with a transepithelial current clamp (Physiologic Instruments, San Diego, CA) and calculating the quotient ISC = Vte/Rte. After the ISC reached a stable plateau (Ibase), amiloride (10 µM, Merck) was applied to the apical chamber. The amiloride-sensitive ISC (∆Iamil), a measure of ENaC activity, was calculated from the difference between Ibase and Iamil. The apical Na^+^ permeability was determined by adding amphotericin B (100 µM, Merck) to the basolateral compartment. For this setup, 140 mM of basolateral Na^+^ was replaced by 116 mM N-methyl-D-glucamine (NMDG^+^, Merck) and 24 mM choline, generating a 145:5 apical-to-basolateral Na^+^ gradient. The ISC was measured every 5 s with a Vte clamp. Following amphotericin B addition, at ISC peak value, amilmax was determined by apically adding 10 µM amiloride, or ouabain (1 mM, Merck) basolaterally to calculate ouabmax.
Western blot
Phosphorylation of Erk1/2 (extracellular-signal-regulated kinases) was analyzed using antibodies against phospho-p44/42 MAPK (Erk1/2) at Tyr202/Tyr204 of Erk1 (Thr185 and Tyr187 of Erk2) (#9101, Cell Signaling Technology, Frankfurt am Main, Germany) and Erk1/2 (#9102, Cell Signaling Technology). Furthermore, α-Tubulin (11H10, #2125, Cell Signaling Technology) expression was used as a reference. Suitable secondary antibodies, conjugated to horseradish peroxidase (HRP), were used to detect primary antibodies. HRP activity was analyzed by enhanced chemiluminescence (ECL, Amersham, Piscataway, NJ, USA) on X-ray film. Band intensity was measured by densitometry using Image-J (National Institutes of Health, Bethesda, MD, USA).
WST-1 assay for cell proliferation and viability
The WST-1 cell proliferation colorimetric assay quantifies cell proliferation, viability, and cytotoxicity by measuring the reduction of water-soluble tetrazolium salt WST-1 to a formazan dye by viable cells and organisms. FDLE cells were seeded in 96-well plates at a density of 2x10^4^ cells per well and infected with Ureaplasma. In parallel, viable Ureaplasma, derived from the same stock, were equally diluted 1:5 in cell culture media. In addition, uninfected FDLE cells were treated with Ureaplasma medium alone, diluted 1:5, serving as negative controls. Then, WST-1 reagent (Roche, Mannheim, Germany) was added to each of the three settings according to the manufacturer’s instructions. Absorbance at 450 nm was measured regularly every 0.5-1 h.
mRNA expression analyses
RNA isolation was done following a 24-h incubation with Ureaplasma using the Purelink RNA Mini Kit (Life Technologies) and DNAse I (Life Technologies) according to the manufacturer’s instructions. For reverse transcription, 1 g of RNA was pre-annealed with Oligo(dT)18 primers (Fisher Scientific GmbH, Schwerte, Germany), followed by the addition of Superscript III (Life Technologies). The Express Greener QPCR Uni-Kit (Fisher Scientific GmbH), gene-specific primers (Table 2), and the CFX 96 Real-Time system (Bio-Rad, Munich, Germany) were used to perform real-time quantitative PCR. Target-specific plasmid DNA was used as the internal standard for absolute quantification. Molecular concentrations were normalized to the mitochondrial ribosomal protein S18a (Mrps18a) reference gene and expressed as the respective control’s relative fold change (RFC).
Table 2: Primer sequences.
Statistical analyses
Statistical analyses were performed using GraphPad Prism software (GraphPad Prism 9.1.1, https://www.graphpad.com, GraphPad Software, La Jolla, CA, USA). Data are presented as means ± standard deviation (SD) and listed in S1 Table. Statistical significance was defined as p-value < 0.05.
Supporting information
S1 FigDetection of pErk1/2 and Erk1/2 in FDLE cells by Western blot analysis.Original blots of Fig 4.(DOCX)
S2 FigDetection of pErk1/2 and Erk1/2 in FDLE cells by Western blot analysis.Original blots of Fig 7A.(DOCX)
S3 FigDetection of pErk1/2 and Erk1/2 in FDLE cells by Western blot analysis.Original blots of Fig 9A.(DOCX)
S1 TableMean, SD and p-values of data.(DOCX)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Lowe J, Watkins WJ, Edwards MO, Spiller OB, Jacqz-Aigrain E, Kotecha SJ, et al. Association between pulmonary ureaplasma colonization and bronchopulmonary dysplasia in preterm infants: updated systematic review and meta-analysis. Pediatr Infect Dis J. 2014;33(7):697–702. doi: 10.1097/INF.0000000000000239 24445836 · doi ↗ · pubmed ↗
- 2Silwedel C, Speer CP, Glaser K. Ureaplasma-associated prenatal, perinatal, and neonatal morbidities. Expert Rev Clin Immunol. 2017;13(11):1073–87. doi: 10.1080/1744666 X.2017.1381559 28918659 · doi ↗ · pubmed ↗
- 3Silwedel C, Laube M, Speer CP, Glaser K. The role of Ureaplasma species in prenatal and postnatal morbidity of preterm infants: current concepts. Neonatology. 2024;121(5):627–35. doi: 10.1159/000539613 38934167 · doi ↗ · pubmed ↗
- 4Viscardi RM, Kallapur SG. Role of Ureaplasma respiratory tract colonization in bronchopulmonary dysplasia pathogenesis: current concepts and update. Clin Perinatol. 2015;42(4):719–38. doi: 10.1016/j.clp.2015.08.003 26593075 PMC 4662049 · doi ↗ · pubmed ↗
- 5Glaser K, Silwedel C, Fehrholz M, Waaga-Gasser AM, Henrich B, Claus H. Ureaplasma species differentially modulate pro- and anti-inflammatory cytokine responses in newborn and adult human monocytes pushing the state toward pro-inflammation. Front Cell Infect Microbiol. 2017;7:484.29234642 10.3389/fcimb.2017.00484 PMC 5712342 · doi ↗ · pubmed ↗
- 6Glaser K, Silwedel C, Fehrholz M, Henrich B, Waaga-Gasser AM, Claus H, et al. Ureaplasma isolates stimulate pro-inflammatory CC chemokines and matrix metalloproteinase-9 in neonatal and adult monocytes. P Lo S One. 2018;13(3):e 0194514. doi: 10.1371/journal.pone.0194514 29558521 PMC 5860755 · doi ↗ · pubmed ↗
- 7Li Y-H, Chen M, Brauner A, Zheng C, Skov Jensen J, Tullus K. Ureaplasma urealyticum induces apoptosis in human lung epithelial cells and macrophages. Biol Neonate. 2002;82(3):166–73. doi: 10.1159/000063616 12373067 · doi ↗ · pubmed ↗
- 8Silwedel C, Fehrholz M, Speer CP, Ruf KC, Manig S, Glaser K. Differential modulation of pulmonary caspases: is this the key to Ureaplasma-driven chronic inflammation? P Lo S One. 2019;14(5):e 0216569. doi: 10.1371/journal.pone.0216569 31067276 PMC 6506144 · doi ↗ · pubmed ↗