Autoimmune Glial Fibrillary Acidic Protein (GFAP) Astrocytopathy Presenting as Viral Encephalitis: A Case Report and Literature Review
Soorya Bavikeri Shivakumara Hegde, Janak Sureshbhai Nayak, Muna Salah, Jeeth Thomas, Brendan Davies

TL;DR
A rare autoimmune condition mimicking viral encephalitis was diagnosed and successfully treated with steroids in a 47-year-old woman.
Contribution
Highlights GFAP astrocytopathy as a rare but treatable cause of encephalitis-like symptoms, emphasizing early immunosuppressive therapy.
Findings
The patient showed significant improvement after high-dose methylprednisolone treatment.
GFAP immunoglobulin G antibodies were detected in serum and cerebrospinal fluid confirming the diagnosis.
Early immunosuppressive therapy improved clinical outcomes in this autoimmune condition.
Abstract
Glial fibrillary acidic protein astrocytopathy is a rare autoimmune condition characterized by antibodies against glial fibrillary acidic protein in astrocytes of the central nervous system. It can present with features of myelitis or meningoencephalitis. Diagnosis is based on the presence of serum or cerebrospinal antibodies against glial fibrillary acidic protein. Most cases respond to immunosuppressive therapy in the form of steroids. A 47-year-old woman with dizziness, tremors, and coryzal symptoms progressed over a period of a few weeks to develop clinical features of brainstem encephalitis. She was started on empirical antimicrobials and anti-convulsive therapy. Although initial MRI showed features of viral encephalitis, this was ruled out after CSF testing, and she was given high-dose methylprednisolone. She was admitted to the intensive care unit in view of her deteriorating…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
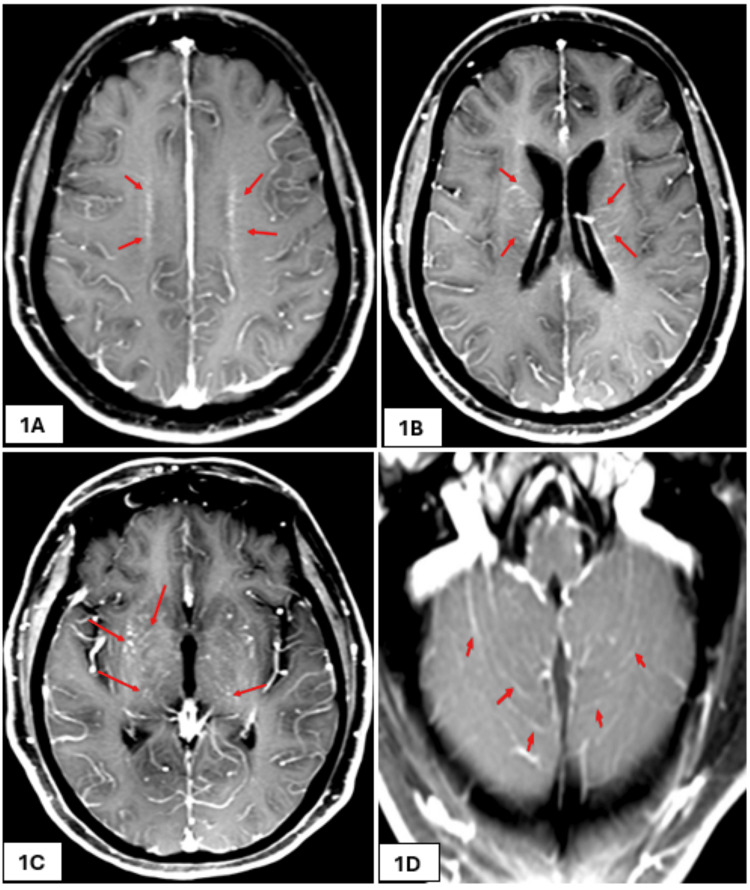 Figure 1
Figure 1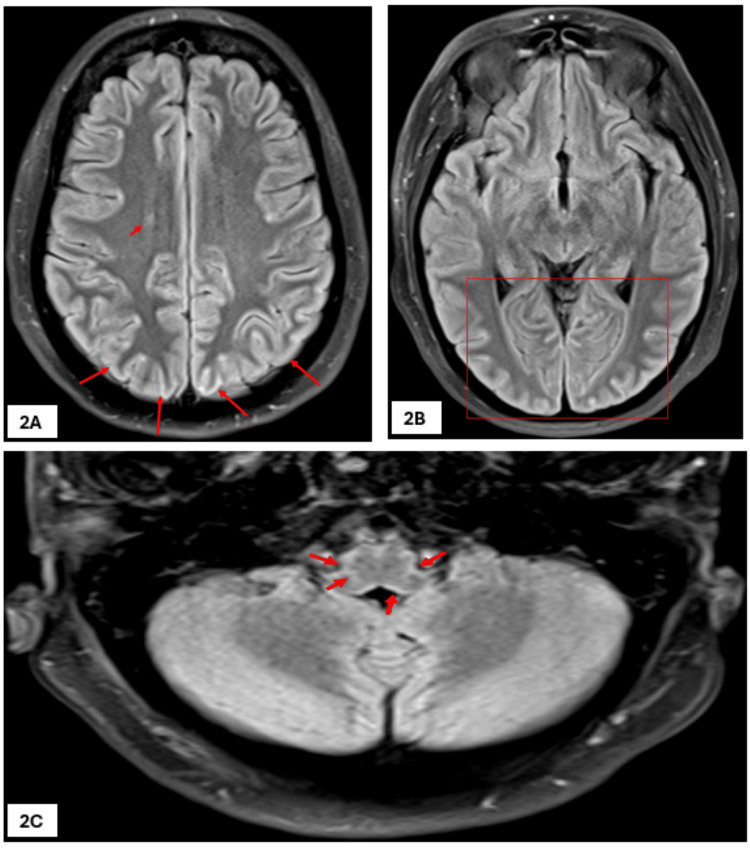 Figure 2
Figure 2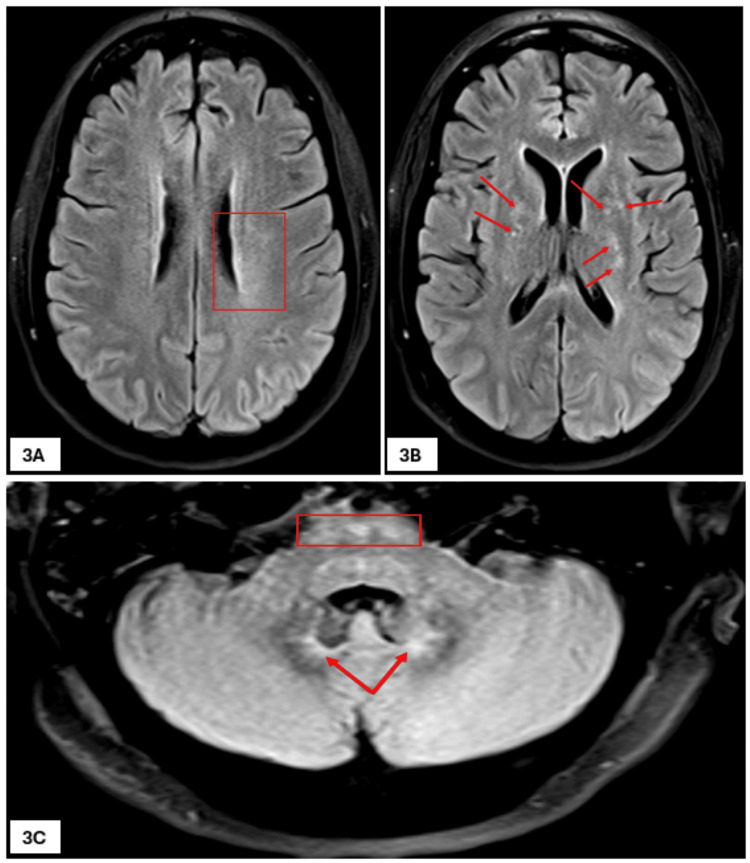 Figure 3
Figure 3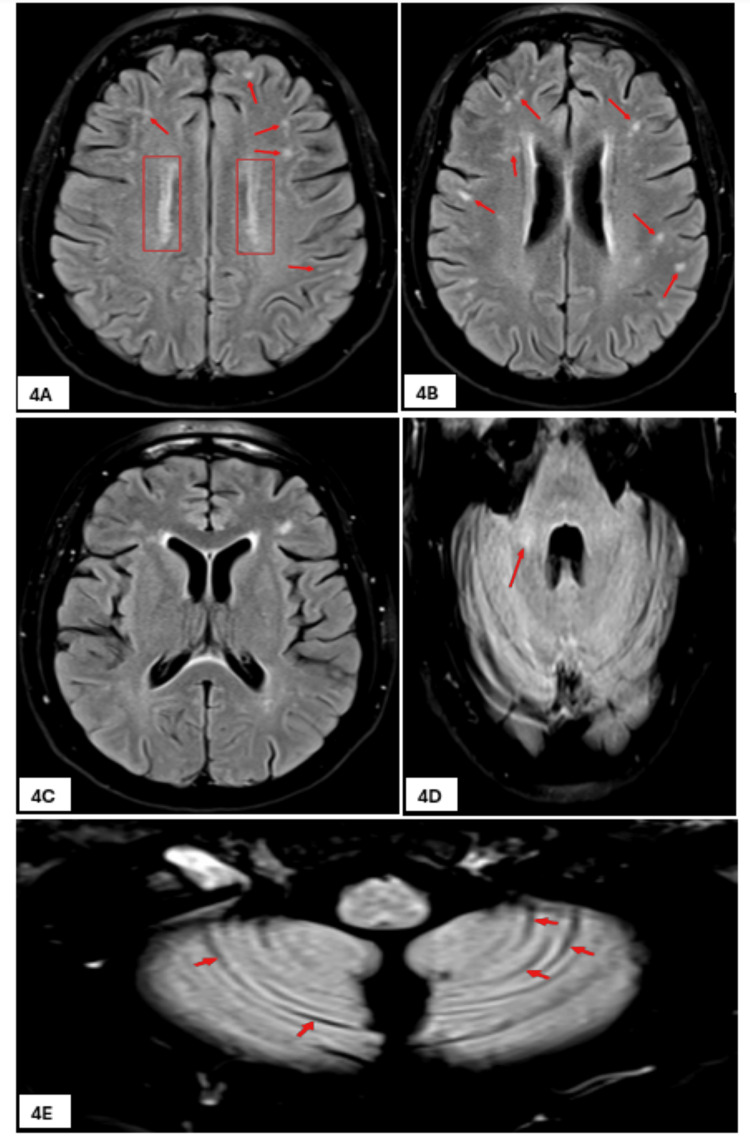 Figure 4
Figure 4| Parameter | First admission to A/E | Second admission to A/E | Reference range |
| Sodium | 131 mmol/L | 137 mmol/L | 133-146 mmol/L |
| Potassium | 3.7 mmol/L | 3.8 mmol/L | 3.5-5.3 mmol/L |
| Urea | 7.6 mmol/L | 14.9 mmol/L | 2.5-7.8 mmol/L |
| Creatinine | 49 mmol/L | 96 mmol/L | 45-84 mmol/L |
| eGFR | >90 mL/min/1.73 m² | 6 mL/min/1.73 m² | >90 mL/min/1.73 m² |
| Magnesium | - | 0.56 mmol/L | 0.70-1.00 mmol/L |
| Inorganic phosphate | - | 0.84 mmol/L | 0.80-1.50 mmol/L |
| Ionized calcium | - | 1.22 mmol/L | 1.16-1.31 mmol/L |
| C-reactive protein (CRP) | - | 8 mg/L | <4 mg/L |
| Albumin | - | 40 g/L | 35-50 g/L |
| Alkaline phosphatase | - | 63 IU/L | 30-130 IU/L |
| Alanine aminotransferase | - | 32 IU/L | 0-34 IU/L |
| Serum bilirubin (total) | - | 26 mmol/L | 0-21 mmol/L |
| Serum ammonia | - | 27 mmol/L | 0-40 mmol/L |
| Hemoglobin | 157 g/L | 157 g/L | 115-165 g/L |
| Red blood cell count | 5.80x1012/L | 5.84x1012/L | 3.80-5.80x1012/L |
| White cell count | 12.50x109/L | 24.60x109/L | 4-11x109/L |
| Lymphocytes | 1.18x109/L | 2.14x109/L | 1.5-4.0x109/L |
| Monocytes | 0.73x109/L | 0.76x109/L | 0.2-0.8x109/L |
| Neutrophils | 10.31x109/L | 21.43x109/L | 2.0-7.5x109/L |
| Platelets | 350x109/L | 512x109/L | 150-450x109/L |
| Parameters | Values | Reference range |
| Erythrocyte sedimentation rate (ESR) | 7 mm/h | 2-16 mm/h |
| Thyroid-stimulating hormone (TSH) | 0.29 mIU/L | 0.38-5.33 mIU/L |
| Triiodothyronine (T3) | 3.8 pmol/L | 3.5-6.5 pmol/L |
| Tetraiodothyronine (T4) | 18.6 pmol/L | 11.5-22.7 pmol/L |
| Thyroid peroxidase (TPO) antibody | Negative | Negative |
| Respiratory pathogens polymerase chain reaction (influenza A, influenza B, COVID-19, and human respiratory syncytial virus) | Not detected | - |
| Procalcitonin | 0.15 µg/L | <0.05 µg/L |
| Legionella and Pneumococcal urinary antigens | Not detected | - |
| Pneumococcal and Meningococcal polymerase chain reaction (PCR) | Negative | - |
| Meningococcal group B polymerase chain reaction (PCR) | Negative | - |
| Multi-resistant Gram-negative bacterial culture screen | Not isolated | - |
| Blood cultures | Staphylococcus hominis | - |
| Sputum culture | No growth | - |
| General bacteriology swab culture (ear swab) | No growth | - |
| Tuberculosis microscopy (direct stain for acid/alcohol fast bacteria) | Not seen | - |
| Ebstein-Barr virus nuclear antigen antibody | IgG positive | - |
| Parvovirus B19 serology | IgG positive | - |
| Cytomegalovirus polymerase chain reaction (PCR) | Not detected | <500 IU/mL |
| BK virus polymerase chain reaction (PCR) | Not detected | - |
| JC virus polymerase chain reaction (PCR) | Not detected | - |
|
| Negative | - |
| Brucella IgM/IgG antibody | <1:20 | <1:20 |
| Anti-extractable nuclear antigen antibodies (includes Ro, La, Smith, RNP, Jo1, and Scl70) | Negative | - |
| Anti-nuclear antibodies | Negative | - |
| Anti-myeloperoxidase antibodies | <0.7 U | <3.5 U |
| Anti-proteinase 3 antibodies | <0.3 U | <2 U |
| Paraneoplastic antibodies | Negative | - |
| Phospholipase A2 receptor antibody | <3.0 RU/mL | 0-13 RU/mL |
| Cardiolipin immunoglobulin G antibody | 0.7 GPL/mL | <10 GPL/mL |
| Glutamic acid decarboxylase (GAD) antibodies | <5.0 IU/mL | 0-10 IU/mL |
| Myelin oligodendrocyte glycoprotein (MOG) antibodies | Negative | - |
| Aquaporin-4 antibodies | Negative | - |
| Acetylcholine receptor antibody | Negative | - |
| Muscle-specific tyrosine kinase IgG antibody | Negative | - |
| Herpes simplex virus 1 and 2 nucleic acid | Not detected | - |
| Varicella zoster virus nucleic acid | Not detected | - |
| HIV-1 and HIV 2 antigens and antibodies | Not detected | - |
| Treponema pallidum antibody screen | Not detected | - |
| Ganglioside antibodies (IgM and IgG) against GM1, GM2, GM3, GM4, GD1a, GD1b, GD2, GD3, GT1a, GT1b, GQ1 | <1:500 (negative) | <1:500 |
| Alpha fetoprotein | <1 kU/L | 0-10 kU/L |
| Angiotensin converting enzyme (ACE) | 28.2 U/L | 13.3-63.9 U/L |
| Schirmer’s test (for Sjogren’s syndrome) | Right eye: 21 mm, left eye: 24 mm | ≥10 mm/5 min |
| Anti-N-methyl-D-aspartate receptor | Negative | - |
| Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor 1 and 2 antibody | Negative | - |
| Dipeptidyl-peptidase-like protein-6 antibodies | Negative | - |
| Contactin-associated protein 2 | Negative | - |
| Leucine-rich glioma-inactivated protein 1 | Negative | - |
| Gamma-aminobutyric acid receptor type B1 | Negative | - |
| Glial fibrillary acidic protein immunoglobulin G (GFAP IgG) | Positive | - |
| Purkinje cell cytoplasmic antibody type 1 (PCA-1/Yo) | Negative | - |
| Anti-neuronal nuclear antibody type 1 (ANNA-1/Hu) | Negative | - |
| Anti-neuronal nuclear antibody type 2 (ANNA-2/Ri) | Negative | - |
| Ma antibody | Negative | - |
| Collapsin response-mediator protein-5/CV2 (CRMP5) antibody | Negative | - |
| Amphiphysin antibody | Negative | - |
| Anti-Sry-like high-mobility group box 1 (SOX1) antibody | Negative | - |
| Glutamic acid decarboxylase (GAD) antibody | Negative | - |
| Zinc-finger protein of the cerebellum 4 (Zic4) antibody | Negative | - |
| Delta/notch-like epidermal growth factor-related receptor (DNER) antibody | Negative | - |
| Parameter | Values (1st sample) | Values (2nd sample) | Reference range |
| Total protein | 1.44 g/L | 0.51 g/L | 0.15-0.45 g/L |
| Glucose | - | 3.8 mmol/L | 2.5-5.5 mmol/L |
| Red blood cells | 88×10⁶/L | 27×10⁶/L | ×10⁶/L |
| White blood cells | 359×10⁶/L | 29×10⁶/L | 0-5×10⁶/L |
| Polymorphs | 0% | 5% | - |
| Mononuclear cells | 100% | 95% | - |
| Organisms | No organisms seen | No organisms seen | - |
| Glial fibrillary acidic protein immunoglobulin G (GFAP IgG) | Not tested | Positive | - |
| Culture | No growth | No growth | - |
| ME film array* | None detected | Not tested | - |
| Anti-N-methyl-D-aspartate receptor | Negative | - | - |
| Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor 1 and 2 antibody | Negative | - | - |
| Dipeptidyl-peptidase-like protein-6 antibodies | Negative | - | - |
| Contactin-associated protein 2 | Negative | - | - |
| Leucine-rich glioma inactivated protein 1 | Negative | - | - |
| Gamma-aminobutyric acid receptor type B1 | Negative | - | - |
| Hospital stay | Course | ICU stay |
| Day 1 | Admitted to the hospital - low GCS with normal CTH. Started on antimicrobials and anti-epileptics | - |
| Day 3 | Transferred to ICU (due to persistent low GCS) 1st EEG (no evidence of epileptiform abnormalities) 1st lumbar puncture | Day 1 |
| Day 4 | Intubated | Day 2 |
| Day 7 | 1st MRI (showing possible post-viral encephalitis) 2nd EEG (no epileptiform abnormalities) | Day 5 |
| Day 11 | Intravenous methylprednisolone started at 1 g/kg/day for 5 days | Day 9 |
| Day 13 | First successful sedation interruption, patient responding to verbal commands, 3rd EEG (no epileptiform abnormalities) | Day 11 |
| Day 14 | Patient is able to follow verbal commands | Day 12 |
| Day 16 | Extubated. Oral prednisolone started at 1 mg/kg/day and was weaned down at 5 mg/week | Day 14 |
| Day 17 | 2nd MRI (post-infective or ischemic changes) | Day 15 |
| Day 21 | Transferred from the ICU to the Neurology unit | Day 19 |
| Day 33 | 3rd MRI (new focal white matter signal abnormalities) | - |
| Day 40 | 2nd lumbar puncture, including samples for glial fibrillary acidic protein antibody testing | - |
| Day 41 | Magnetic resonance angiogram (MRA) (unremarkable) | - |
| Day 48 | Discharged from the hospital | - |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAutoimmune Neurological Disorders and Treatments · Peripheral Neuropathies and Disorders · Brain Metastases and Treatment
Introduction
Glial fibrillary acidic protein (GFAP) astrocytopathy was first described in 2016 [1]. It is a rare autoimmune condition that can involve various parts of the central nervous system (CNS), like the subcortical white matter, brainstem, medulla, hypothalamus, basal ganglia, or spinal cord. An incidence of 0.03 per 100,000 person-years and a prevalence of 0.6 per 100,000 people have been reported in a previous study [2]. GFAP astrocytopathy has been reported in both adults and children. It is more commonly seen in middle-aged adults, with a slight male preponderance [3].
Inflammation from certain immune cells (lymphocytes and granulocytes), support cells (astrocytes), and the complement system has been implicated in the pathogenesis [4]. This results in features of meningitis, encephalitis, or myelitis, like fever, headache, abnormal vision, altered consciousness, seizures, movement disorders, hyperreflexia, and ophthalmoplegia [5]. Due to its similarity to infections, such as viral encephalitis, it is often misdiagnosed or initially mistaken for an infectious process. The relationship between viral infections and GFAP astrocytopathy is still an area of active research. A recent meta‐analytical study revealed that 45% of patients had a viral prodrome, primarily upper respiratory or gastrointestinal infections, with some rarer cases involving HIV and bacterial infections [3]. The time between the appearance of viral symptoms and the onset of neurological manifestations is usually brief, typically occurring within weeks to months [3]. Although it is part of the autoimmune encephalitis spectrum, it is distinct in its pathophysiology and clinical presentation, and is characterized by specific immune reactions involving glial fibrillary acidic protein immunoglobulin G antibodies (GFAP-IgG) in cerebrospinal fluid (CSF) [6]. Magnetic resonance imaging can show typical findings, such as periventricular radial and linear contrast enhancement [7,8], and treatment involves steroids or intravenous immunoglobulin therapy [9].
We present a case of a middle-aged woman where the diagnosis was protracted due to the clinico-radiological resemblance to infectious encephalitis. We also emphasize the importance of early consideration of autoimmune mechanisms in the diagnostic process and the prompt initiation of appropriate therapy to improve patient outcomes.
Case presentation
Initial presentation (admission days one and two)
A 47-year-old Caucasian woman with a history of type 2 diabetes mellitus was initially seen in the general practice for dizziness, for which she was given prochlorperazine 5 mg tablets (to take as required). When her symptoms worsened, she presented to the Accident and Emergency (A&E) department. Neurological examination was unremarkable, and blood tests were as shown in Table 1. She was given cyclizine 50 mg tablets for vestibular neuritis and discharged. She then developed worsening dizziness, tinnitus, poor balance, and new-onset tremors with no other neurological symptoms. This was put down to the side effects of cyclizine therapy and treated with betahistine 16 mg tablets. She was re-admitted to A/E with worsening dizziness, tremors, loss of balance, agitation, and hallucinations. The patient was unable to stand due to ataxia and tremor, with an initial Glasgow Coma Scale (GCS) of 15/15. Blood tests showed raised infection markers and acute kidney injury (Table 1). A panel of tests done as part of a full septic screen, to rule out other causes of infection, were unremarkable. Following specialist input obtained from the neurology and infectious diseases team, she was treated along the lines of meningoencephalitis with 2 g of intravenous (IV) ceftriaxone twice a day, 10 mg/kg of IV acyclovir three times a day, and 500 mg of IV metronidazole given three times a day. These antimicrobials were given for a total of 14 days. She was also given 3 g of intravenous levetiracetam and 2 g of IV sodium valproate as a loading dose, followed by 1.5 g of levetiracetam twice a day and 600 mg of sodium valproate three times a day intravenously for maintenance to cover non-convulsive status epilepticus. She was given sedation for agitation and started on variable rate insulin infusion due to the risk of impending diabetic ketoacidosis (DKA). GCS had now dropped to 3, and computed tomography (CT) of the brain showed no intracranial abnormalities to explain the drop in her GCS.
ICU course (admission days three to 17)
On initial assessment by the intensive care unit (ICU) team, her GCS had improved to 9/15. She was maintaining her airway and following commands. On further review, there was no improvement in her GCS, and she was noted to have clonus of the upper limbs. She was admitted to the ICU after intubation under sedation. CT venogram of the cerebral veins showed no evidence of cerebral sinus venous thrombosis, and magnetic resonance imaging (MRI) showed diffuse perivascular leptomeningeal enhancement in a posterior circulation distribution with minor parenchymal swelling, as shown in Figures 1A-1D, 2A-2C, which was suggestive of post-viral, perhaps post-COVID, encephalitis, as per the neuroradiologist. Once infective causes were included, she was started on 1 g methyl prednisolone per day intravenously for five days. A dramatic improvement was noted in her clinical status within two days of starting IV steroids. This was followed by a weaning dose of oral prednisolone tablets, which was started at 1 mg/kg body weight and reduced at a rate of 5 mg/week as per the advice from the neurologist. An electroencephalogram (EEG) done at this point showed a burst suppression pattern with no clear intermixed epileptic discharges.
First MRI scan of brain (FAT-SAT axial images).Post-contrast T1 fat saturation (FAT-SAT) axial images showing bilateral symmetrical increased perivascular enhancement along the medullary veins - best appreciated at corona radiata level (long arrows in 1A) as well as basal ganglia and midbrain regions (long arrows in 1B and 1C) together with diffuse leptomeningeal enhancement extending to the scanned upper cervical cord (short arrows in 1D).
First MRI scan of brain (FLAIR images).Two-dimensional axial fluid-attenuated inversion recovery (FLAIR) images showing right frontal deep periventricular focus of FLAIR signal elevation (short arrow in 2A) with subtle cortical swelling and elevated FLAIR signal seen along high parietal (long arrows in 2A), posterior temporal and occipital polar regions (rectangle in 2B), and medulla oblongata (short arrows in 2C).
Over the next two weeks in the ICU, she was managed by a multidisciplinary team consisting of a neurologist, a microbiologist, a rheumatologist, and an infectious diseases specialist. Clinically, she had convergent strabismus, head version to the left, initial anisocoria, involuntary myoclonus, asymmetric action tremors evolving into visual hallucinations, rhythmic hyperkinetic movements of her left shoulder, and bilaterally positive Hoffman’s sign. This was clinically suspected to indicate subacute brainstem encephalitis. Sodium valproate was now weaned down at the rate of 300 mg per day and then stopped. An extensive set of investigations was conducted to identify the cause of her presentation, with the results presented in Table 2. The results of the first lumbar puncture were unremarkable and are presented in Table 3.
Two further EEGs performed within a span of seven days did not show any evidence of epileptiform abnormalities. A second MRI of the brain showed progressive development of bilateral deep periventricular white matter and basal ganglia foci of elevated fluid-attenuated inversion recovery (FLAIR) signal (Figures 3A-3C). MRI of the spine, however, did not show any significant findings. CT of thorax, abdomen, and pelvis showed bilateral incidental pulmonary embolism during this stay, for which she was initially started on treatment dose of dalteparin given subcutaneously once a day and later changed to apixaban 10 mg tablets taken twice a day for seven days, followed by 5 mg tablets twice a day for maintenance.
Second MRI scan of the brain.Two-dimensional axial FLAIR images showing progressive changes - bilateral deep periventricular white matter with indistinct areas of FLAIR signal alteration (rectangle in 3A); basal ganglia foci of elevated FLAIR signal (arrows in 3B); indistinct foci in the pontine tegmentum (rectangle in 3C); and the ventral aspects of both dentate nuclei (arrows in 3C).FLAIR: fluid-attenuated inversion recovery
Recovery (admission days 21-48)
At the end of her two-week stay in the ICU, she was extubated and transferred to care under the neurology team for neurorehabilitation and monitoring. She was treated by a multidisciplinary team of dieticians, speech and language therapists, diabetes team, occupational therapists, and physiotherapists. Her speech and cognition improved, and clinical examination revealed bilateral proximal weakness in all four limbs with no sensory loss and symmetric action tremors, which gradually improved during inpatient stay. She was able to mobilize with the aid of a frame, and there were no significant pyramidal or extrapyramidal signs noted at this point. An ultrasound scan of the parotid glands, performed to rule out Sjogren’s syndrome as per rheumatology input, showed parotid glands that were normal in size, shape, and parenchymal echotexture. A third MRI of the brain showed newly developed focal subcortical and deep white matter high signal intensities predominantly involving the supratentorial brain with resolution of basal ganglia foci as well as tegmental pontine signal alteration (Figures 4A-4E).
Third MRI scan of the brain.Two-dimensional axial fluid-attenuated inversion recovery (FLAIR) images showing progressive development of bilateral subcortical foci (arrows in 4A and 4B) and deep periventricular white matter sheets of FLAIR signal elevation (rectangle in 4A), with the latter showing predilection for a perimedullary venous location (4A and 4B compared with 1A, 2A, and 3A), with resolution of the basal ganglia foci (4C compared with 3B) as well as tegmental pontine signal alteration (4D dcompared with 3C), and interval development of a right middle cerebellar peduncle ill-defined focus of FLAIR signal alteration (arrow in 4D compared with 3C), and development of early cerebellar atrophic changes (arrows in 4E compared with 1D).
This was presumed to be due to a vasculitic or inflammatory process by the neuro-radiology multidisciplinary team, especially in view of a previous pulmonary embolus. Blood testing done previously also included tests to rule out vasculitis (Table 2). Magnetic resonance angiography (MRA) was done in view of the same, which did not show any features of vasculitis. Fundoscopic examination revealed a normal fundus, visual field, visual acuity, and color vision. Repeat CSF testing showed a downward trend in mononuclear cells and the presence of GFAP IgG antibodies (Table 3). These antibodies were also found in her serum, which helped formulate the final diagnosis (Table 2). She was discharged on a weaning dose of prednisolone and continues to make good progress under the care of the community rehabilitation team. Further review has been arranged with the neurology team. A timeline of key events during her hospital and ICU stay is presented in Table 4.
Discussion
Brainstem encephalitis or rhombencephalitis is a rare infectious or inflammatory condition affecting parts of the brainstem. The diagnosis can be challenging and is commonly caused by infections like Listeria [10], although there have been reports of post-viral brainstem encephalitis secondary to COVID-19 infection [11], with some cases secondary to inherited deficiency of the RNA lariat-debranching enzyme 1 (DBR1), as described by Chan et al. in their study [12].
Glial fibrillary acidic protein (GFAP) astrocytopathy is a rare autoimmune condition characterized by antibodies in the blood or cerebrospinal fluid against GFAP alpha. It is thought to be due to immune reactions involving macrophages, plasma cells, helper T cells [13], and cytotoxic T cells [4]. It is associated with viral infections like COVID-19 or malignancies like breast cancer [14,15]. There are rare instances of the coexistence of GFAP astrocytopathy and myelin oligodendrocyte glycoprotein immunoglobulin G (MOG-IgG) disease [16]. Although there are no agreed diagnostic criteria, a few typical features, such as linear perivascular radial enhancement or leptomeningeal enhancement on imaging, and the presence of IgG GFAP antibodies in the CSF, can aid in diagnosis [17]. Onset can be subacute, and the clinical course severe, with features of brain-stem involvement. Treatment primarily involves immunosuppression with steroids [11]. The long-term outcomes seem to be favorable, although relapses can be seen in 20-30% of the cases with higher rates associated with concurrent NMDAR-IgG antibodies, AQP-4 antibodies [18], and tumor at the onset of symptoms [17,18].
In the present case, the patient had multiple initial visits to the emergency department with tremors and dizziness and progressed over a course of a few weeks to develop encephalopathic features with brainstem involvement. She was started on anticonvulsive and antimicrobial therapy and extensively investigated by a multidisciplinary team, and the cause of her presentation was initially not known. An excellent response to corticosteroid therapy and MRI findings of radial perivascular enhancement indicated a possible autoimmune pathophysiology of the encephalitis [6,8]. Potential differentials included GFAP astrocytopathy, anti-NMDAR encephalitis, MOG antibody disease, and neuromyelitis optica spectrum disorder (NMOSD). Repeat CSF testing near the end of her hospital stay revealed IgG antibodies to glial fibrillary acidic protein in both the CSF and the serum. Neuronal antibodies for other autoimmune encephalitides were negative (Table 2). As illustrated in the present report, diagnosis can be challenging due to the possibility of concurrent presentation or cases mimicking CNS infections, with a subsequent risk of misdiagnosis as intracranial infections [19-22]. Potential consequences include the administration of inappropriate treatment and subsequent delays in initiating proper therapy, which may adversely affect patient outcomes. This underscores the significance of keeping a wider array of differentials and starting early empirical therapy.
After discharge, the patient experienced some initial cognitive challenges, including memory and attention difficulties, which are common in GFAP astrocytopathy. Over time and with rehabilitation, she showed significant improvement, particularly in verbal memory and problem-solving. However, mild cognitive slowing and attention issues persisted. She continues to make good progress under rehabilitation and is on a weaning dose of prednisolone. Further follow-up has been arranged.
Conclusions
Glial fibrillary acidic protein (GFAP) astrocytopathy is a rare autoimmune condition affecting the central nervous system, characterized by the presence of immunoglobulin G antibodies against GFAP alpha in the serum or cerebrospinal fluid. It can present clinically and radiologically with features of infectious encephalitis and can pose a diagnostic challenge due to a protracted clinical course. It is important to consider autoimmune pathophysiology in cases of infectious encephalitis and to conduct relevant investigations, including CNS imaging, serological testing, and neurophysiological studies, at first instance. Empirical initiation of corticosteroid-based immunosuppression at an early stage has been suggested to reduce illness severity and improve patient outcomes, consistent with the existing literature.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Autoimmune glial fibrillary acidic protein astrocytopathy: a review of the literature Front Immunol Shan F Long Y Qiu W 9201810.3389/fimmu.2018.02802 PMC 629089630568655 · doi ↗ · pubmed ↗
- 2Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis Ann Neurol Dubey D Pittock SJ Kelly CR 1661778320182929327310.1002/ana.25131 PMC 6011827 · doi ↗ · pubmed ↗
- 3Clinical and neuroimaging phenotypes of autoimmune glial fibrillary acidic protein astrocytopathy: a systematic review and meta-analysis Eur J Neurol Hagbohm C Ouellette R Flanagan EP 31202410.1111/ene.16284 PMC 1123575138506182 · doi ↗ · pubmed ↗
- 4New insights into neuropathology and pathogenesis of autoimmune glial fibrillary acidic protein meningoencephalomyelitis Acta Neuropathol Guo Y Endmayr V Zekeridou A 147202410.1007/s 00401-023-02678-7PMC 1083824238310187 · doi ↗ · pubmed ↗
- 5Glial fibrillary acidic protein autoimmunity: a French cohort study Neurology Gravier-Dumonceau A Ameli R Rogemond V 65366898202210.1212/WNL.0000000000013087 PMC 882996334799461 · doi ↗ · pubmed ↗
- 6Autoimmune glial fibrillary acidic protein astrocytopathy Curr Opin Neurol Kunchok A Zekeridou A Mc Keon A 4524583220193072476810.1097/WCO.0000000000000676 PMC 6522205 · doi ↗ · pubmed ↗
- 7Unveiling distinctive MRI characteristics in the diagnosis of GFAP astrocytopathy: a rare autoimmune neuroinflammatory disorder Ann Indian Acad Neurol Charan BD Priya S Goel V Chhatarpal P Jain S Gupta A Garg A 3163182720243885616010.4103/aian.aian_1134_23PMC 11232841 · doi ↗ · pubmed ↗
- 8MRI findings of autoimmune glial fibrillary acidic protein astrocytopathy involving infratentorial: case report Radiol Case Rep Ma W Huang C Yang L Luo J 251525181720223560137810.1016/j.radcr.2022.04.032PMC 9114157 · doi ↗ · pubmed ↗