Incidence, Risk Factors, and Treatment of Autoimmune Cytopenia Following Pediatric Allogeneic Hematopoietic Stem Cell Transplantation
Changlan Chen, Yingying Wang, Yan Meng, Ying Dou, Luying Zhang, Xianmin Guan, Xiaoying Lei, Jie Yu

TL;DR
This study examines autoimmune cytopenia in children after stem cell transplants, identifying risk factors and treatment outcomes.
Contribution
The study identifies chronic graft-versus-host disease as an independent risk factor for autoimmune cytopenia in pediatric stem cell transplant patients.
Findings
37 out of 436 pediatric patients developed autoimmune cytopenia after allogeneic stem cell transplantation.
Chronic graft-versus-host disease was identified as an independent risk factor for autoimmune cytopenia.
Steroids and intravenous immunoglobulin achieved complete remission in nearly half of the patients with autoimmune cytopenia.
Abstract
Autoimmune cytopenia (AIC) following pediatric allogeneic hematopoietic stem cell transplantation (allo‐HSCT) is relatively rare but it is a challenging complication, and standardized treatment guidelines are lacking. We retrospectively analyzed 436 pediatric patients undergoing allo‐HSCT; 37 (8.5%) developed AIC, characterized by autoimmune hemolytic anemia (n = 13), immune thrombocytopenia (n = 11), and Evans syndrome (n = 13). Risk factor analysis revealed that younger age at HSCT, nonmalignant diseases, unrelated donor transplantation, and chronic graft‐versus‐host disease (cGVHD) were significantly associated with the development of AIC. Through multivariate analysis, cGVHD was identified as an independent risk factor for AIC. In our study, the first‐line treatment for AIC involved steroids and/or intravenous immunoglobulin, with a complete remission rate of 48.6%. Additional…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
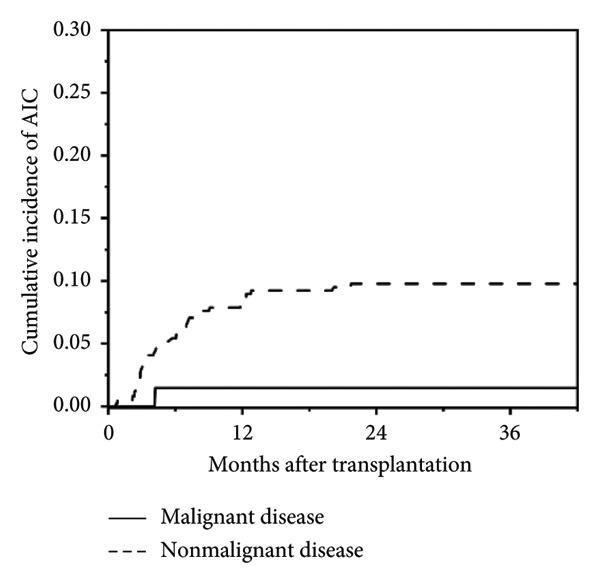 Figure 1
Figure 1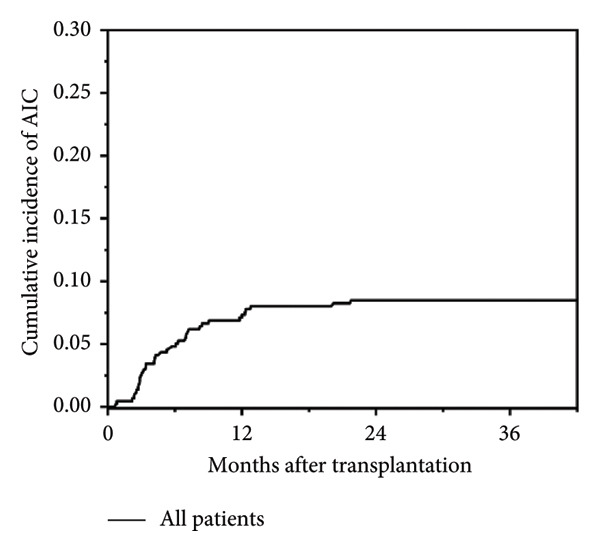 Figure 2
Figure 2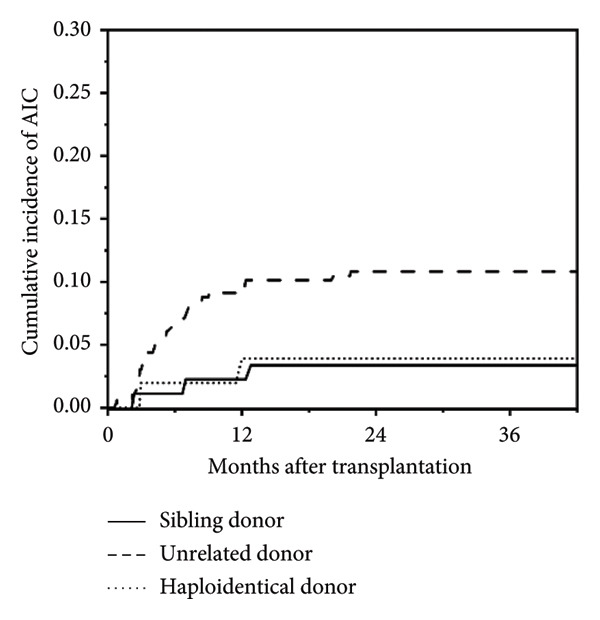 Figure 3
Figure 3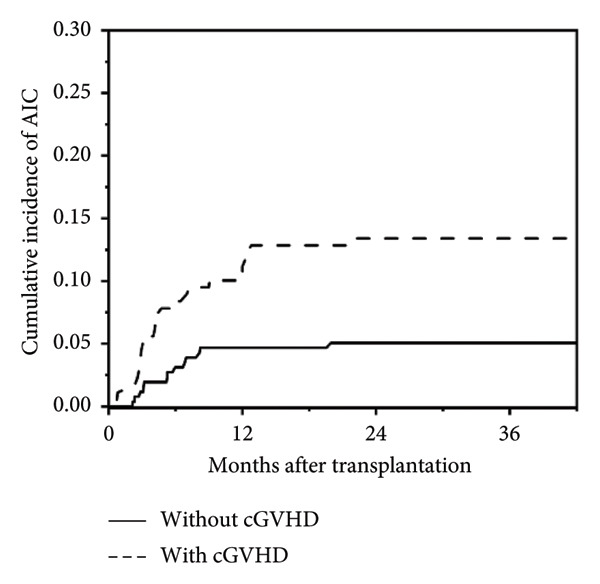 Figure 4
Figure 4| Characteristics | Patients with AIC | Patients without AIC |
|
|---|---|---|---|
| No. of patients | 37 | 399 | |
| Median age at HSCT, year (range) | 2.1 (0.7–17.4) | 3.3 (0.3–14.8) | 0.028 |
| Gender | |||
| Male | 29 (78.4) | 276 (69.2) | 0.243 |
| Female | 8 (21.6) | 123 (30.8) | |
| Diagnosis | |||
| Malignant | 1 (2.7) | 67 (16.8) | 0.024 |
| Nonmalignant | 36 (97.3) | 332 (83.2) | |
| Donor type | |||
| Sibling donor | 3 (8.1) | 86 (21.6) | 0.040 |
| Unrelated donor | 32 (86.5) | 264 (66.2) | |
| Haploidentical donor | 2 (5.4) | 49 (12.3) | |
| Stem cell source | |||
| Bone marrow∗ | 0 | 14 (3.5) | 0.105 |
| Peripheral blood | 33 (89.2) | 369 (92.5) | |
| Cord blood | 4 (10.8) | 16 (4.0) | |
| Serotherapy | |||
| ATG | 35 (94.6) | 331 (83.0) | 0.065 |
| No ATG | 2 (5.4) | 68 (17.0) | |
| HLA match | |||
| 10/10 | 17 (45.9) | 196 (49.1) | 0.712 |
| ≤ 9/10 | 20 (54.1) | 203 (50.9) | |
| ABO donor/recipient | |||
| Identical | 11 (29.7) | 158 (39.6) | 0.358 |
| Compatible (donor O+) | 6 (16.2) | 73 (18.3) | |
| Other | 20 (54.1) | 168 (42.1) | |
| CMV/EBV viremia | |||
| EBV | 12 (32.4) | 107 (26.8) | 0.800 |
| CMV | 1 (2.7) | 27 (6.8) | |
| Both | 20 (54.1) | 222 (55.6) | |
| Acute GVHD, any | 30 (81.1) | 285 (71.4) | 0.210 |
| Grades I‐II | 26 (70.3) | 238 (59.6) | |
| Grades III‐IV | 4 (10.8) | 47 (11.8) | |
| Chronic GVHD, any | 24 (64.9) | 155 (38.8) | < 0.001 |
| Limited | 17 (46.0) | 136 (34.1) | |
| Extensive | 7 (18.9) | 19 (4.7) | |
| Chimerism | |||
| Full | 32 (86.5) | 344 (86.2) | 0.321 |
| Mixed | 5 (13.5) | 29 (7.3) | |
| Graft failure | 0 | 14 (3.5) | |
| untested# | 0 | 12 (3.0) |
| Risk factors | Odds ratio | 95% CI |
|
|---|---|---|---|
| Age at HSCT (year) | 0.881 | 0.769–1.010 | 0.069 |
| Diagnosis | |||
| Nonmalignant vs. malignant | 5.465 | 0.706–42.277 | 0.104 |
| Donor | |||
| URD vs SD | 2.742 | 0.799–9.414 | 0.109 |
| HID vs SD | 0.666 | 0.098–4.548 | 0.679 |
| cGVHD | |||
| Limited vs. absent | 2.392 | 1.110–5.156 | 0.026 |
| Extensive vs. absent | 9.868 | 3.161–30.811 | 0.000 |
| No. | Diagnosis | Sex | Age at HSCT, yr(s) | Age at AIC, yr(s) | HSCT type | Chimerism status | aGVHD maximum grade | cGVHD | AIC | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Donor | Stem source | Conditioning regimen | |||||||||
| 1 | ALL | F | 2.1 | 2.4 | MMUD | CB | BU + CY | Full | III | Limited | ITP |
| 2 | WAS | M | 1.0 | 1.4 | MUD | CB | BU + CY | Full | II | Limited | AIHA |
| 3 | Thalassemia M | M | 1.2 | 1.8 | MMUD | PB | BU + FLU + ATG | Full | II | Limited | AIHA + ITP |
| 4 | WAS | M | 3.6 | 3.8 | MMUD | CB | BU + FLU + ATG | Full | I | Limited | AIHA + ITP |
| 5 | SCID | M | 17.4 | 17.6 | MSD | PB | BU + FLU + ATG | Full | Absent | Limited | ITP |
| 6 | WAS | M | 1.7 | 1.9 | MMUD | CB | BU + FLU + ATG | Full | II | Absent | ITP |
| 7 | Thalassemia M | M | 1.5 | 2.1 | MMSD | PB | CY + BU + FLU + ATG | Full | II | Absent | AIHA |
| 8 | WAS | M | 0.9 | 1.6 | MUD | PB | BU + CY + ATG | Full | II | Absent | AIHA |
| 9 | Thalassemia M | M | 1.9 | 3.8 | MMUD | PB | CY + BU + FLU + ATG | Full | II | Limited | ITP |
| 10 | CGD | M | 0.8 | 1.1 | MUD | PB | BU + CY + ATG | Full | I | Absent | ITP |
| 11 | CGD | M | 0.9 | 1.4 | MMUD | PB | BU + CY + ATG | Full | II | Absent | AIHA + ITP |
| 12 | WAS | M | 0.7 | 1.0 | MMUD | PB | BU + CY + ATG | Mixed | II | Limited | ITP |
| 13 | Thalassemia M | M | 2.0 | 3.0 | MMUD | PB | BU + CY + FLU + ATG | Full | Absent | Limited | AIHA |
| 14 | CGD | M | 0.8 | 1.4 | MUD | PB | BU + CY + ATG | Full | I | Absent | AIHA |
| 15 | CGD | M | 7.5 | 8.6 | MSD | PB | BU + CY + ATG | Full | II | Extensive | ITP |
| 16 | CGD | M | 5.0 | 5.2 | MMUD | PB | BU + CY + ATG | Mixed | Absent | Absent | AIHA |
| 17 | WAS | M | 1.6 | 2.5 | MMUD | PB | BU + CY + ATG | Full | II | Limited | AIHA |
| 18 | HIGM | M | 3.5 | 3.9 | MUD | PB | BU + CY + ATG | Full | I | Absent | AIHA + ITP |
| 19 | HIGM | M | 1.9 | 2.4 | MUD | PB | BU + CY + ATG | Full | Absent | Absent | AIHA |
| 20 | WAS | M | 0.8 | 1.6 | MMUD | PB | BU + CY + ATG | Full | II | Extensive | AIHA + ITP |
| 21 | CGD | M | 1.3 | 1.9 | MUD | PB | BU + CY + ATG | Full | I | Absent | AIHA |
| 22 | CGD | M | 2.7 | 3.1 | MUD | PB | BU + CY + ATG | Full | I | Limited | AIHA + ITP + AIN |
| 23 | Thalassemia M | M | 2.3 | 2.5 | MUD | PB | CY + BU + FLU + ATG | Mixed | I | Absent | AIHA |
| 24 | Thalassemia M | F | 2.1 | 3.8 | MUD | PB | CY + BU + FLU + ATG | Full | Absent | Absent | AIHA |
| 25 | Thalassemia M | F | 6.2 | 6.4 | MUD | PB | CY + BU + FLU + ATG | Full | II | Absent | ITP |
| 26 | SAA | F | 11.1 | 11.3 | MUD | PB | CY + BU + FLU + ATG | Full | II | Limited | ITP |
| 27 | HIGM | M | 1.4 | 1.7 | MUD | PB | BU + CY + ATG | Full | II | Limited | AIN + AIHA |
| 28 | Thalassemia M | M | 1.8 | 2.4 | MUD | PB | CY + BU + FLU + ATG | Mixed | Absent | Limited | AIHA |
| 29 | Thalassemia M | F | 3.1 | 3.3 | MUD | PB | CY + BU + FLU + ATG | Full | I | Extensive | AIHA + ITP + AIN |
| 30 | HIGM | M | 3.3 | 3.3 | MMUD | PB | CY + BU + ATG | Full | III | Limited | AIHA + ITP |
| 31 | Thalassemia M | F | 2.4 | 3.4 | MMUD | PB | CY + BU + FLU + ATG | Full | II | Limited | ITP |
| 32 | WAS | M | 6.3 | 6.6 | MMUD | PB | CY + BU + ATG | Full | II | Extensive | ITP |
| 33 | Thalassemia M | M | 4.8 | 5.0 | MMUD | PB | CY + BU + FLU + ATG | Full | II | Limited | ITP + AIN |
| 34 | Thalassemia M | F | 2.8 | 3.5 | MMUD | PB | CY + FLU + BU + ATG | Full | II | Extensive | AIHA + ITP |
| 35 | SAA | F | 4.5 | 4.8 | HID | PB | CY + BU + FLU + ATG | Full | III | Extensive | AIHA + ITP + AIN |
| 36 | WAS | M | 1.2 | 2.2 | HID | PB | BU + CY + ATG | Mixed | Absent | Limited | AIHA |
| 37 | Thalassemia M | M | 2.5 | 2.6 | MMUD | PB | CY + FLU + BU + ATG | Full | IV | Extensive | AIHA + ITP |
| No. | Type of AIC | Time to AIC, d | Time to CR, d | Treatment and response | Outcome | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| First‐line therapy | Response | Second‐line therapy | Response | Third‐ line therapy | Response | |||||
| 1 | ITP | 126 | 35 | MPD | CR | N/A | N/A | N/A | N/A | Alive |
| 2 | AIHA | 128 | 119 | MPD + IVIG | CR | N/A | N/A | N/A | N/A | Alive |
| 3 | AIHA + ITP | 189 | 341 | MPD + IVIG | NR | PX ∗ 3 + CY ∗ 2 + RTX ∗ 4 | CR | N/A | N/A | Alive |
| 4 | AIHA + ITP | 102 | 133 | MPD | CR | N/A | N/A | N/A | N/A | Alive |
| 5 | ITP | 70 | 145 | PDN | CR | N/A | N/A | N/A | N/A | Alive |
| 6 | ITP | 74 | 119 | PDN | CR | N/A | N/A | N/A | N/A | Alive |
| 7 | AIHA | 209 | 68 | MPD | CR | N/A | N/A | N/A | N/A | Alive |
| 8 | AIHA | 253 | 281 | MPD | NR | MPD + IVIG + RTX ∗ 4 | CR | N/A | N/A | Alive |
| 9 | ITP | 652 | 81 | PDN | CR | N/A | N/A | N/A | N/A | Alive |
| 10 | ITP | 96 | 86 | MPD + IVIG | CR | N/A | N/A | N/A | N/A | Alive |
| 11 | AIHA + ITP | 172 | 37 | MPD + IVIG | CR | N/A | N/A | N/A | N/A | Alive |
| 12 | ITP | 142 | NR | MPD + IVIG | NR | N/A | N/A | N/A | N/A | Dead |
| 13 | AIHA | 371 | 15 | MPD | CR | N/A | N/A | N/A | N/A | Alive |
| 14 | AIHA | 214 | 163 | MPD | NR | RTX ∗ 5 + IVIG | CR | N/A | N/A | Alive |
| 15 | ITP | 384 | 152 | MPD + IVIG | CR | N/A | N/A | N/A | N/A | Alive |
| 16 | AIHA | 65 | 273 | MPD | CR | N/A | N/A | N/A | N/A | Alive |
| 17 | AIHA | 354 | 931 | MPD | NR | PDN + MMF | PR | MMF + SRL | CR | Alive |
| 18 | AIHA + ITP | 158 | 53 | MPD | CR | N/A | N/A | N/A | N/A | Alive |
| 19 | AIHA | 182 | 31 | MPD | CR | N/A | N/A | N/A | N/A | Alive |
| 20 | AIHA + ITP | 272 | NR | IVIG + PDN | NR | MPD + RTX ∗ 1 | NR | N/A | N/A | Dead |
| 21 | AIHA | 247 | 16 | MPD + IVIG | CR | N/A | N/A | N/A | N/A | Alive |
| 22 | AIC | 124 | 117 | MPD + IVIG | NR | MPD + RTX ∗ 2 | CR | N/A | N/A | Alive |
| 23 | AIHA | 102 | 74 | MPD + IVIG | NR | MPD + RTX ∗ 6 | NR | SRL + MMF | CR | Alive |
| 24 | AIHA | 606 | PR | IVIG + PDN | PR | N/A | N/A | N/A | N/A | Alive |
| 25 | ITP | 86 | NR | MPD | NR | RTX ∗ 1 | NR | N/A | N/A | Dead |
| 26 | ITP | 77 | 108 | MPD | CR | N/A | N/A | N/A | N/A | Alive |
| 27 | AIN + AIHA | 86 | NR | MPD + IVIG | NR | SRL | NR | N/A | N/A | Dead |
| 28 | AIHA | 210 | PR | MPD | NR | PDN + MMF | PR | N/A | N/A | Alive |
| 29 | AIC | 92 | PR | MPD + IVIG | NR | RTX ∗ 5 | NR | SRL + MMF | PR | Dead |
| 30 | AIHA + ITP | 24 | 105 | MPD | NR | RTX ∗ 4 | NR | SRL | CR | Alive |
| 31 | ITP | 368 | 76 | MPD + IVIG | CR | N/A | N/A | N/A | N/A | Alive |
| 32 | ITP | 81 | 39 | MPD + IVIG | NR | PDN + TPO | CR | N/A | N/A | Alive |
| 33 | ITP + AIN | 83 | NR | MPD | NR | RTX ∗ 1 | NR | N/A | N/A | Dead |
| 34 | AIHA + ITP | 218 | NR | IVIG + PDN | NR | MMF + PDN | NR | SRL | NR | Dead |
| 35 | AIC | 88 | 54 | MPD | NR | RTX ∗ 1 | CR | N/A | N/A | Dead |
| 36 | AIHA | 360 | 71 | MPD | CR | N/A | N/A | N/A | N/A | Alive |
| 37 | AIHA + ITP | 22 | NR | MPD | NR | RTX ∗ 4 | NR | SRL | NR | Dead |
- —Specialized Research Project on Stem Cell Therapy
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlatelet Disorders and Treatments · Blood disorders and treatments · Hematopoietic Stem Cell Transplantation
1. Introduction
Autoimmune cytopenia (AIC) following allogeneic hematopoietic stem cell transplantation (allo‐HSCT) is a relatively rare yet serious complication, characterized by the destruction of donor‐derived blood cells mediated by the donor’s immune system, leading to singular or multilineage cytopenia. Autoimmune hemolytic anemia (AIHA) is the predominant manifestation, followed by immune thrombocytopenia (ITP), Evans’ syndrome, and autoimmune neutropenia (AIN). AIC predominantly occurs within 5–10 months post‐HSCT, with an incidence of 5%–10% in pediatric patients [1–6], and surging up to 20% in specific nonmalignant diseases [7, 8]. Reported risk factors include younger age at transplantation, nonmalignant diseases, unrelated donor transplantation, cord blood transplantation, the use of lymphocyte‐depleting agents in the conditioning regimen, chronic graft‐versus‐host disease (cGVHD), and viral infection [5, 9–13]. Underlying mechanisms may be rooted in incomplete immune reconstitution or immune dysregulation post‐HSCT. This therapeutic approach for post‐HSCT AIC predominantly aligns with strategies established for primary AIHA. First‐line treatment includes steroids and/or intravenous immunoglobulin (IVIG), yielding remission in a majority of patients [5, 14]. However, recurrence rates are notably elevated, particularly upon tapering steroids. Additional treatment options include rituximab (RTX), sirolimus [2, 4, 5, 10], and newer immunosuppressive therapies, such as bortezomib [15, 16], daratumumab [17, 18], and abatacept [19], which have shown promising results in some patients.
The management of post‐transplant AIC remains challenging, as responses to immunosuppressive therapy are often incomplete or nondurable [4]. In addition, post‐transplant AIC is often associated with other complications, such as immunodeficiency, infections, and GVHD. The concurrence of these conditions not only increases diagnostic and therapeutic complexity but also exacerbates immune dysregulation, leading to a significantly increased risk of mortality [9, 20]. Despite the introduction of targeted biological agents and supportive care advances, the prognosis for severe or refractory post‐transplant AIC remains unsatisfactory [15, 17], highlighting the urgent need for more effective therapeutic strategies.
This retrospective study examined clinical data from 436 pediatric patients undergoing allo‐HSCT at the Hematology‐Oncology Center of Chongqing Medical University Affiliated Children’s Hospital between January 2014 and December 2021. It aimed to assess the incidence and risk factors and describe treatment outcomes for patients with post‐transplant AIC. The findings may provide guidance for the management and treatment of refractory/recurrent AIC.
2. Patients and Methods
2.1. Patients
This study was approved by the Institutional Review Board of Children’s Hospital of Chongqing Medical University (ethics approval number: 2023 Lunshen [Research] No. 36). Because this is a retrospective study, the need for patient consent for data collection was waived by the Institutional Review Board. This study involved 436 pediatric patients who underwent allo‐HSCT at the Hematology‐Oncology Center of Chongqing Medical University Affiliated Children’s Hospital from January 2014 to December 2021. The patients were categorized into two groups: one with malignant diseases, such as acute leukemia and myelodysplastic syndrome, and the other with nonmalignant diseases, including severe thalassemia, severe aplastic anemia, lymphoproliferative diseases, and primary immunodeficiency diseases (e.g., Wiskott–Aldrich syndrome, chronic granulomatous disease, severe combined immunodeficiency, hyper‐IgM syndrome, and others). Patients who underwent allo‐HSCT and subsequently developed AIC were included for analysis. Before transplantation, all patients were screened using the direct antiglobulin test (DAT). Patients with DAT positivity due to ABO antibodies, history of AIHA, or positive DAT results before HSCT were excluded from our study. Conditioning regimens are tailored as either myeloablative or reduced intensity based on the primary disease. Post‐transplantation, GVHD prophylaxis included cyclosporine, mycophenolate mofetil, and/or methotrexate. All patients received IVIG support therapy initiated on day 7 post‐transplant at a dose of 0.4–0.5 g/kg weekly, followed by monthly infusions during outpatient follow‐up for approximately 1 year, until immune reconstitution was achieved.
2.2. AIC Definitions and Diagnosis
AIHA was diagnosed based on the following criteria: (1) hemoglobin < 100 g/L (or a significant decrease from baseline) with a reticulocyte count > 120 × 10^9^/L or > 3%; (2) laboratory evidence of hemolysis, including lactate dehydrogenase > 1.5 × upper limit of normal (ULN), total bilirubin > 1.5 × ULN, and haptoglobin < 0.3 g/L; (3) positive DAT; and (4) exclusion of patients with DAT positivity mediated by ABO antibodies, a history of AIHA, or prior DAT positivity before allo‐HSCT, as well as other causes of immune hemolytic anemia. ITP and AIN pertain to unexplained isolated thrombocytopenia or neutropenia, defined by a platelet count < 100 ∗ 10^9^/L and a neutrophil count < 1.0 ∗ 10^9^/L, respectively. Other potential causes of cytopenia, such as drug toxicity, GVHD, or underlying viral infections following HSCT, were ruled out. In appropriate patients, bone marrow biopsy was performed to exclude cytopenia attributed to bone marrow insufficiency. Evans syndrome refers to the simultaneous or sequential occurrence of cytopenia involving two or three cell lineages.
2.3. Treatments
In our study, first‐line therapy for AIC consisted of steroids, initiated at 4–6 mg/kg/day and escalated to 10–20 mg/kg/day in severe cases as clinically indicated, followed by gradual tapering according to therapeutic response. Adjunctive interventions, including IVIG (0.5–1 g/kg/day), transfusion support, and other supportive measures, were applied based on clinical need. Patients refractory to first‐line therapy received rituximab at 375 mg/m^2^ weekly for 4–6 doses. Sirolimus, at 0.5–2 mg/day, was subsequently introduced if additional immunosuppression was required. In patients with ITP, thrombopoietin was administered to enhance platelet production. Further therapy adjustments were guided by patient response and the discretion of the attending physicians.
Treatment response was defined as follows: complete response, normalization of hemoglobin (age‐adjusted for children), a platelet count > 100 × 10^9^/L, and a neutrophil count > 1.5 × 10^9^/L, accompanied by normalization of hemolytic markers and sustained transfusion independence for at least 2 months; partial response, hemoglobin increase > 2 g/dL without transfusion in the preceding 7 days, accompanied by improvement in hemolytic markers, a platelet count at least twice the pretreatment level without bleeding, and a neutrophil count > 1.0 × 10^9^/L, with continued therapy to maintain the response; and nonresponse, unchanged or worsening manifestations despite treatment.
2.4. Post‐HSCT Monitoring Parameters
For patients undergoing allo‐HSCT, cytomegalovirus (CMV) and Epstein–Barr virus (EBV) loads were monitored weekly during the first 2 months post‐HSCT using quantitative polymerase chain reaction (PCR). Subsequently, monitoring continued during all scheduled outpatient clinic visits. The diagnostic thresholds for viral viremia were defined as CMV > 1000 copies/mL and EBV > 2000 copies/mL.
Chimerism analysis was performed on days +14 after HSCT and continued monthly until stabilization, using short tandem repeat (STR)–PCR technology. Complete chimerism was defined as > 95% of donor chimerism in peripheral blood. Mixed chimerism was defined as between 5% and 95% of donor chimerism in peripheral blood, or in one or more lineage. Graft failure was characterized by donor chimerism < 5%. Cases of early mortality post‐HSCT were categorized under unassessed STR status due to the inability to conduct subsequent evaluations.
2.5. Statistical Analysis
Baseline characteristics of the study cohort were summarized using descriptive statistics. Quantitative variables, represented as median and range, underwent univariate analysis through analysis of the Mann–Whitney U test. Categorical variables were presented as absolute number and percentage, and univariate analysis was performed using the chi‐square test or Fisher’s exact test. The following variables were included in the univariate analysis of factors predicting the development of AIC: gender, primary diseases, median age at HSCT, donor type, stem cell source, the use of ATG serotherapy, ABO and HLA compatibility, acute and chronic GVHD, CMV or EBV viremia, and chimerism status at the time of AIC diagnosis. Variables with p < 0.05 in univariate analysis were subjected to multivariate logistic regression analysis to identify independent risk factors for post‐HSCT AIC. The endpoints were the incidence of AIC and treatment response. Statistical analyses were performed using the SPSS software Version 25.0. All p values were 2‐sided, and p < 0.05 was considered statistically significant. p > 0.1 was reported as not significant, whereas p between 0.05 and 0.1 was reported in detail.
3. Results
3.1. Patients
In this study, a total of 436 patients underwent allo‐HSCT, with a median age at transplantation of 3.3 years (range: 0.3–17.4 years) and a median follow‐up duration of 21.8 months (range: 0.1–102.4 months). Among them, 37 patients (8.5%) developed post‐transplant AIC, including AIHA (n = 13), ITP (n = 11), and Evans syndrome (n = 13, including 8 AIHA + ITP, 1 AIN + AIHA, 1 AIN + ITP, and 3 AIHA + ITP + AIN). No isolated AIN was observed. Cumulative incidence of post‐transplant AIC is shown in Figure 1. The median age at onset of post‐transplant AIC was 2.6 years (range: 1.0–17.6 years), and the median time to onset was 147 days post‐transplant (range: 22–652 days). Detailed baseline HSCT characteristics of all patients are summarized in Table 1.
Figure 1. Cumulative incidence of post‐transplant AIC. (a) All patients; (b) patients with malignant disease versus patients with nonmalignant disease. (c) Patients with sibling donor versus patients with unrelated donor versus patients with haploidentical donor; (d) patients with cGVHD versus patients without cGVHD.(a)(b)(c)(d)
3.2. Risk Factors
Univariate analysis showed no statistically significant differences between patients with and without AIC in terms of gender, stem cell source, ATG serotherapy, HLA and ABO compatibility, EBV/CMV viremia, chimerism status, or acute GVHD occurrence. However, significant associations with AIC were revealed in the following variables: younger age at HSCT (2.1 years [range, 0.7–17.4] in patients with AIC vs. 3.3 years [range, 0.3–14.8 years] in those without AIC; p = 0.028), primary diagnosis of a nonmalignant disease (p = 0.024), unrelated donor transplantation (p = 0.040), and concurrent cGVHD (p < 0.001). Multivariate analysis further delineated cGVHD as the only independent risk factor for AIC development post‐HSCT, as detailed in Table 2.
3.3. Treatment and Outcomes
Transplant characteristics for AIC patients are summarized in Table 3, and the treatment and outcomes for these individuals are detailed in Table 4. All patients received steroids and/or IVIG as first‐line treatment, supplemented with transfusion therapy and plasma exchange as clinically required, achieving a complete remission rate of 48.6% (18/37). Other second‐line or more advanced therapeutic approaches involved RTX (n = 12), sirolimus (n = 7), mycophenolate mofetil (n = 5), thrombopoietin (n = 1), and cyclophosphamide (n = 1). Of these, RTX achieved complete remission in 5 of the 12 patients treated. Sirolimus resulted in complete remission for 3 of 7 patients, and thrombopoietin successfully induced complete remission in the single patient treated. Overall, 27 patients (73.0%) reached complete remission, with a median remission duration of 86 days (range, 15–931 days). Additionally, 3 patients (8.1%) experienced partial remission.
At the last follow‐up, 28 of the AIC patients (75.7%) were alive, while 9 (34.3%) died. Among these, one patient with AIHA, ITP, and AIN achieved partial remission but died due to a severe infection and immunological encephalitis on day 351. Another patient, despite reaching complete remission for AIHA + ITP + AIN, died due to complications of immunological encephalitis and impaired consciousness. The causes of death for the others included severe aspergillosis in 3 patients and multiorgan bleeding in 4.
4. Discussion
This study presents the incidence, risk factors, and treatments for AIC after allo‐HSCT. The median time to onset was 147 days post‐transplant (range: 22–652 days), with an incidence of 8.5% (37/436), aligning with findings from previous studies [1, 8, 21]. Notably, 36 of 37 patients had nonmalignant diseases, in particular they were patients with high IgM syndrome, where the incidence of AIC post‐transplantation reached 26.7% (4/15). Recent studies have indicated that nonmalignant diseases are associated with the development of AIC after transplantation. For instance, Page et al. reported a 56% incidence of AIC in children with metabolic diseases post‐transplant [22], and Kruizinga et al. found that β‐thalassemia was present in 9 of 26 post‐transplant AIC patients [10]. This trend suggests that pretransplant abnormal immune status in patients with nonmalignant diseases may elevate the risk for developing autoimmune disorders post‐transplant.
Our study indicates that risk factors for post‐transplant AIC include younger age at transplantation, nonmalignant diseases, unrelated donor transplantation, and concurrent cGVHD. Notably, cGVHD has been identified as an independent risk factor for the development of AIC. Studies, such as Lum et al., have also underscored the role of cGVHD in the development of AIC [5], suggesting that it negatively affects immune reconstitution and thymic regeneration, potentially leading to regulatory T‐cell deficiency and increased AIC susceptibility [23]. Patients with extensive gastrointestinal cGVHD warrant special attention due to the increased severity and complexity of their condition. For example, patients 20, 34, and 37 developed AIHA and ITP after HSCT, accompanied by severe gastrointestinal cGVHD, resulting in significant anemia, infections, diarrhea, and multiorgan bleeding. Even with the use of multiple drugs in combination, disease control was challenging, ultimately leading to the mortality of these 3 patients.
Additionally, existing research points to potential correlations between AIC development and factors, such as cord blood transplantation [6, 11, 14] and viral infections, notably CMV [10]. Yet, our findings do not conclusively validate these associations. This could be due to the relatively small number of patients involving cord blood transplantation (4.6%) and the high prevalence of concurrent CMV and/or EBV infections in the patients (89.2%). Despite this, careful monitoring for viral infections, particularly CMV, remains crucial due to its implicated role in immune system dysregulation [10, 24].
The underlying pathogenesis of post‐transplant AIC is not fully understood, possibly associated with incomplete immune reconstitution or immune dysregulation after transplantation. Some studies suggest that the use of immunosuppressive agents (such as alemtuzumab or ATG) pre‐HSCT to suppress T‐cell proliferation may result in an imbalance in lymphocyte subsets, potentially promoting autoimmune reactions [2, 6, 10]. Notably, in our study, 94.6% (35/37) of AIC patients received ATG serotherapy (p = 0.065). Among patients who underwent immune assessments at the time point closest to AIC onset, we observed that B‐cell recovery occurred more rapidly than T‐cell reconstitution in most patients, suggesting that early B‐cell predominance may contribute to immune imbalance and AIC development. A small proportion of patients, however, did not undergo complete immune profiling, which limited our ability to comprehensively evaluate the relationship between immune reconstitution and post‐transplant AIC. Nevertheless, these observations may serve as a useful guide for future research exploring the pathogenesis of AIC.
Existing research indicates that the majority of AIC patients occur in the setting of complete chimerism, suggesting that the autoantibodies against donor blood cells stem from donor plasma cells [10, 20, 25, 26]. However, the study by Even‐Or et al. revealed that 46.2% (6/13) of patients exhibited mixed chimerism at the time of AIC diagnosis, implying that residual antibody‐secreting host cells could also play a role in its pathogenesis [1]. This may potentially account for the 5 patients of mixed chimerism observed in our study.
In our study, the first‐line treatment included steroids and/or IVIG, achieving a complete remission rate of 48.6%, a higher rate compared to most reported studies [5, 14, 27]. When first‐line treatment is ineffective, second‐ or third‐line therapies, such as RTX and sirolimus, are commonly employed to suppress aberrant immune responses. Both these drugs have been proven effective in prior research [4, 5, 14]. Our study highlights that sirolimus was typically administered in the mid‐ to late stages of the disease when patients often presented with severe complications, such as severe GVHD and infections. This complexity could potentially impact the expected efficacy of sirolimus. Therefore, early consideration of combination therapies to control disease progression is crucial when managing refractory or recurrent AIC. Additional treatments employed in our study, such as mycophenolate mofetil and cyclophosphamide, have been documented in other studies [4, 11]; yet, these approaches yielded limited therapeutic efficacy.
In our study, 9 AIC patients (24.3%) died, sharing a common characteristic of abnormal immune status (such as immunodeficiency, severe GVHD, and other autoimmune disorders). Additionally, the increased risk of bleeding due to thrombocytopenia further complicated matters. The interaction of these factors made patients more susceptible to severe infections, autoimmune responses, and multiorgan bleeding. Therefore, exploring innovative therapeutic strategies is crucial for effectively managing and halting the progression of AIC. Recent studies have highlighted the efficacy of bortezomib, daratumumab, and abatacept in treating refractory or recurrent AIC [18, 19, 28–30]. These drugs could be worth considering for future treatments.
5. Conclusion
Our study revealed an incidence of AIC after allo‐HSCT of 8.5%, notably higher among patients with nonmalignant diseases, with concurrent cGVHD identified as a significant independent risk factor. First‐line treatment involving steroids and/or IVIG resulted in a 48.6% complete remission rate, while the follow‐up treatments, such as RTX and sirolimus, also helped some patients to achieve complete remission. Overall, the treatment of post‐transplant AIC is challenging and usually associated with poor prognosis, the severity of which may vary from individual to individual, and sometimes may even lead to death. Therefore, close monitoring of patients who may develop AIC is required when allo‐HSCT is performed, especially those with nonmalignant disease and severe cGVHD. For patients who have already developed AIC, the necessary therapeutic measures need to be taken in a timely manner to avoid adverse outcomes.
Disclosure
All authors approved the final manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Author Contributions
Changlan Chen and Jie Yu conceptualized the study. Changlan Chen, Yingying Wang, Yan Meng, and Xiaoying Lei participated in data collection and statistical analysis. Changlan Chen and Yingying Wang drafted the manuscript. Ying Dou, Luying Zhang, Xianmin Guan and Xiaoying Lei, and Jie Yu critically reviewed the draft. Changlan Chen, Yingying Wang, and Yan Meng revised the manuscript.
Funding
The authors thank the support of the Specialized Research Project on Stem Cell Therapy (No. 30000222).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Even-Or E. , Schejter Y. D. , Nasereddin A. , Zaidman I. , Shadur B. , and Stepensky P. , Autoimmune Cytopenias Post Hematopoietic Stem Cell Transplantation in Pediatric Patients With Osteopetrosis and Other Nonmalignant Diseases, Frontiers in Immunology. (2022) 13, 10.3389/fimmu.2022.879994.PMC 918513735693771 · doi ↗ · pubmed ↗
- 2Neely J. A. , Dvorak C. C. , Pantell M. S. , Melton A. , Huang J. N. , and Shimano K. A. , Autoimmune Cytopenias in Pediatric Hematopoietic Cell Transplant Patients, Frontiers in Pediatrics. (2019) 7, 171–179, 10.3389/fped.2019.00171, 2-s 2.0-85064623799.31131266 PMC 6509944 · doi ↗ · pubmed ↗
- 3Miller P. D. E. , Snowden J. A. , De Latour R. P. et al., Autoimmune Cytopenias (AIC) Following Allogeneic Haematopoietic Stem Cell Transplant for Acquired Aplastic Anaemia: A Joint Study of the Autoimmune Diseases and Severe Aplastic Anaemia Working Parties (ADWP/SAAWP) of the European Society for Blood and Marrow Transplantation (EBMT), Bone Marrow Transplantation. (2020) 55, no. 2, 441–451, 10.1038/s 41409-019-0680-4, 2-s 2.0-85074027836.31554929 PMC 6995778 · doi ↗ · pubmed ↗
- 4Koo J. , Giller R. H. , Quinones R. , Mc Kinney C. M. , Verneris M. R. , and Knight‐Perry J. , Autoimmune Cytopenias Following Allogeneic Hematopoietic Stem Cell Transplant in Pediatric Patients: Response to Therapy and Late Effects, Pediatric Blood and Cancer. (2020) 67, no. 9, 28591–28602, 10.1002/pbc.28591.32658382 · doi ↗ · pubmed ↗
- 5Lum S. H. , Selvarajah S. , Deya-Martinez A. et al., Outcome of Autoimmune Cytopenia After Hematopoietic Cell Transplantation in Primary Immunodeficiency, Journal of Allergy and Clinical Immunology. (2020) 146, no. 2, 406–416, 10.1016/j.jaci.2020.04.053.32442647 · doi ↗ · pubmed ↗
- 6Szanto C. L. , Langenhorst J. , De Koning C. et al., Predictors for Autoimmune Cytopenias After Allogeneic Hematopoietic Cell Transplantation in Children, Biology of Blood and Marrow Transplantation. (2020) 26, no. 1, 114–122, 10.1016/j.bbmt.2019.07.022.31344451 · doi ↗ · pubmed ↗
- 7Horn B. , Viele M. , Mentzer W. , Mogck N. , De Santes K. , and Cowan M. , Autoimmune Hemolytic Anemia in Patients With SCID After T Cell-Depleted BM and PBSC Transplantation, Bone Marrow Transplantation. (1999) 24, no. 9, 1009–1013, 10.1038/sj.bmt.1702011, 2-s 2.0-0032714360.10556961 · doi ↗ · pubmed ↗
- 8Deambrosis D. , Lum S. H. , Hum R. M. et al., Immune Cytopenia Post–Cord Transplant in Hurler Syndrome is a Forme Fruste of Graft Rejection, Blood Advances. (2019) 3, no. 4, 570–574, 10.1182/bloodadvances.2018026963, 2-s 2.0-85061989278.30787020 PMC 6391675 · doi ↗ · pubmed ↗