The role of the TIM-3 receptor in inflammatory bowel disease and autoimmunity: implications for TIM-3 and TCR crosstalk
M. Gabel, A. Knauss, M. F. Neurath, B. Weigmann

TL;DR
This review explores how the TIM-3 receptor interacts with T cell receptors to regulate immune responses, focusing on its role in inflammatory bowel disease and autoimmunity.
Contribution
The paper provides a comprehensive overview of TIM-3's molecular interactions and signaling in the context of TCR activation and autoimmune diseases.
Findings
TIM-3 interacts with TCR signaling through kinases like ITK and Lck to regulate T cell activation.
Phosphorylation of TIM-3 by ITK and displacement of Bat3 modulate early TCR signaling.
TIM-3's role in inflammatory bowel disease and autoimmunity is highlighted with open questions outlined.
Abstract
The T cell immunoglobulin and mucin-domain receptor 3 (TIM-3) is an immune checkpoint receptor with complex effects on T cell activation and tolerance. TIM-3 is expressed on the surface of T cells in close proximity to the T cell receptor (TCR), where it mainly acts co-inhibitory, despite lacking typical inhibitory immunoreceptor inhibition motifs, to regulate T cell activity or apoptosis. However, the underlying molecular mechanisms of its intracellular signal transduction remains incompletely understood. Central for the function of TIM-3 are conserved cytoplasmic tyrosine structures that require phosphorylation for downstream signaling. Previous findings suggest that several kinases linked to TCR signaling, including the Src family kinase Lck and the Tec kinase ITK can interact with TIM-3 and thereby regulate TCR signaling and fine-tune T cell activation and tolerance. The…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
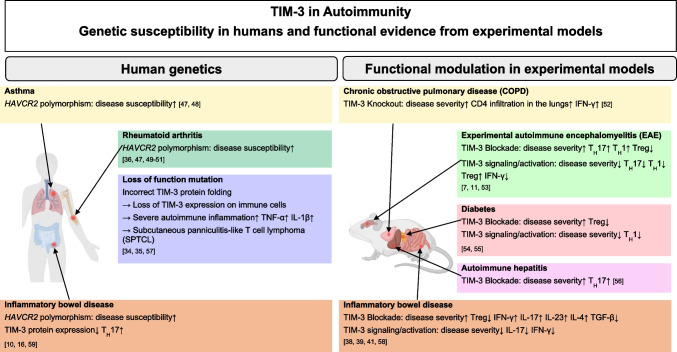 Figure 1
Figure 1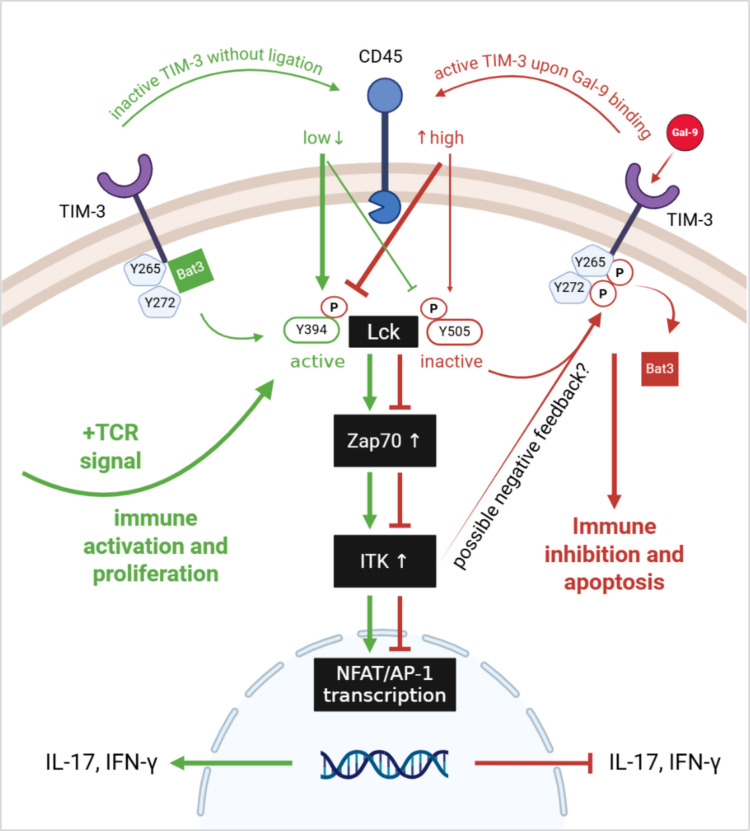 Figure 2
Figure 2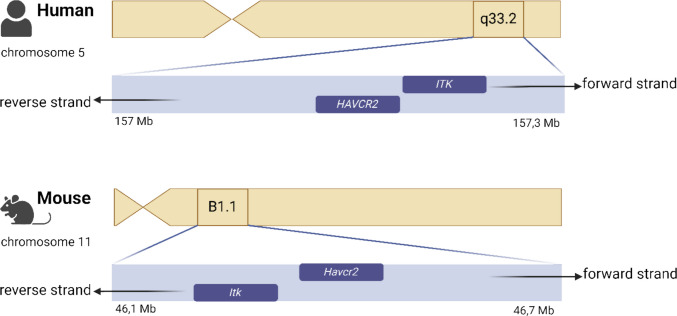 Figure 3
Figure 3- —Universitätsklinikum Erlangen (8546)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGalectins and Cancer Biology · Signaling Pathways in Disease · Cancer Mechanisms and Therapy
Introduction
T cells are essential players of the adaptive immune system providing effective immune responses [1, 2]. Upon immune stimulation, antigen-presenting cells facilitate T cell activation by presenting foreign antigens on their surface, to which the T cell receptor (TCR) can bind. The subsequent signal is transmitted into the cell via a complex biochemical process involving molecular changes, with the participation of several proteins and kinases at the cell membrane, but also in the cytoplasm. Downstream TCR signaling cascades result in the activation of transcription factors that initiate the transcription and translation of genes into proteins which enables T cell effector functions and differentiation processes [3, 4]. Additional receptors on the T cell surface can have a co-stimulatory or co-inhibitory effect on TCR signaling, thereby modulating intracellular signal transduction and amplifying or dampening T cell activity [5]. When excessive or chronic antigen stimulation occurs, co-inhibitory receptors are increasingly expressed on T cells, which in turn dampen excessive T cell activity and the associated damage caused by overactive T cell responses [6]. The T cell immunoglobulin and mucin-domain receptor 3 (TIM-3) is a known immune checkpoint receptor with identified co-inhibitory functions [7]. Recently, this receptor attracted great interest not only in context with cancer but also due to its involvement in the pathogenesis of autoimmune diseases and particularly in inflammatory bowel disease (IBD) [7–10]. Early studies established a well-known role of TIM-3 in mediating apoptosis of T cells, especially T_H_1 cells, upon binding to its ligand galectin-9 making TIM-3 popular for dampening T_H_1 immune responses [11]. Nevertheless, individual studies attribute a dual role for TIM-3 and also describe co-stimulatory functions of TIM-3, which often leads to discussions about if TIM-3 is a co-inhibitory or co-stimulatory receptor [12]. These uncertainties originate partially from specific structural features of the TIM-3 receptor. The TIM-3 receptor has an immunoglobulin variable domain, a mucin domain, a transmembrane domain, and an intracellular cytoplasmic tail. Unlike other better-studied immune checkpoint receptors, such as PD-1, it has no known inhibitory immunoreceptor tyrosine-based inhibition motifs (ITIMs) in the region of its cytoplasmic tail [13]. However, TIM-3 has six tyrosine residues within its cytoplasmic tail, of which two tyrosines can be phosphorylated for downstream signaling [14–16]. Although several studies have attempted to investigate the function of TIM-3 and its precise signaling, many questions remain unanswered, particularly in relation to its interaction with the T cell receptor downstream cascade. The effects and functions of TIM-3 are highly dependent on the phosphorylation of its cytoplasmic tyrosine structures, which can be mediated by several TCR-related kinases [15]. This review aims to summarize current findings in TIM-3 signaling, its interconnectivity to TCR signaling, and its linkage to autoimmune disorders with an emphasis on IBD. Furthermore, key players of TIM-3’s intracellular phosphorylation will be highlighted and gaps in knowledge identified raising open questions for future research.
Characteristics of the TIM-3 molecule and its ligands
The gene of TIM-3 is termed Hepatitis A virus cellular receptor 2 (HAVCR2) in humans and Havcr2 in mice, respectively [17]. HAVCR2 RNA expression shows high levels in the human brain, kidney and urinary bladder, as well as in hematopoietic and lymphoid tissues, particularly in lymph nodes and spleen (data obtained from the human protein atlas: https://www.proteinatlas.org [18]). Lymphoid tissues exhibit higher TIM-3 protein expression compared to most other organ tissues. Comparable TIM-3 protein expression is observed in the gastrointestinal tract, whereas the highest TIM-3 protein expression is detected in the kidney [18]. RNA single cell data specifically identified NK cells, macrophages, monocytes, and granulocytes as relevant immune cells expressing HAVCR2. However, in colon tissue T cells display the highest HAVCR2 transcript [18]. Pioneering work from Monney et al., identified and characterized the TIM-3 protein as a T_H_1-specific cell surface protein consisting of 281 amino acids in mice and 301 amino acids in humans [7]. They further detected a 63% amino acid identity between the human and murine protein and located their genes on the human chromosome 5.q33 and mouse chromosome 11B1 [7]. The protein is composed of an immunoglobulin variable-region-like domain and a mucin domain consisting of serine and threonine residues by 31%. Both domains represent the extracellular compound of the protein providing sites for both O-linked and N-linked glycosylation. The extracellular domains are followed by a transmembrane domain and an intracellular region containing 6 tyrosine residues [7, 19]. In mice the residues Y256 (corresponding to Y265 in human TIM-3 receptor) and Y263 (corresponding to Y272 in human TIM-3 receptor) are reported to be of special interest as they enable interactions with the human leukocyte antigen B (HLA-B) associated transcript 3 (Bat3) [14, 20], more on this later. In 2007, Cao et al. identified and described unique structural features of the murine TIM-3 protein. Its IgV domain varies from other immunoglobulin superfamily members as it shapes a so-called FG-CC cleft. Two additional intramolecular disulfide bonds are formed by four noncanonical cysteines resulting in close proximity of the CC’ loop to the FG’ loop that are located across each other in classical IgV domains [21].
At present, four ligands have been discovered for the TIM-3 proteins namely galectin-9 (Gal-9), phosphatidylserine (PtdSer), the high-mobility group box 1 (HMGB1) as well as the carcinoembyrotic antigen-related cell adhesion molecule 1 (CEACAM1). They are presented in the following section.
Gal-9 (encoded by the LGALS9 gene) was the first TIM-3-ligand identified by Zhu et al. [22]. It is a carbohydrate-recognizing protein, belongs to the galectin family, also known as S-type lectins, can be found in both humans and mice, and has a molecular mass of 30–35 kD [23]. Gal-9 is widely expressed in the organ systems and by various cell types. High protein levels can be found including but not limited to the brain, lung, and gastrointestinal tract whereas RNA levels are highly increased in the blood [24]. Single cell analysis identified enterocytes, paneth cells and goblet cells as major cell types expressing LGALS9 [18]. Upon binding to the extracellular TIM-3 glycosylation sites, Gal-9 can provoke T_H_1 cell death in vitro within 4–8 h associated with a distinct calcium flux; no such effect was detected in T_H_2 cells [11]. However, apoptosis of T_H_1 cells was not fully eradicated in TIM-3-deficient cells proposing that Gal-9 may mediate cell death by binding additional receptors. Zhu et al. further reported that Gal-9 can eliminate IFN-γ producing CD4^+^T cells and reviewed that Gal-9 expression can be stimulated by IFN-γ and IL-1β. This suggests a negative feedback loop in which IFN- γ can not only promote inflammatory processes but can also induce inhibitory Gal-9 counteracting and dampening inflammation by targeting T_H_1 cells via TIM-3 [11].
PtdSer is a phospholipid and part of plasma membranes [25]. PtdSer binds to the structural pocket formed by the CC and FG loops of the TIM-3 protein and all other proteins of the TIM family except for murine TIM-2 [26]. PtdSer is a marker designating cells that undergo apoptosis. In healthy cells, it is localised within the plasma membrane but it is exposed on the cell surface of apoptotic cells in order to be recognized by phagocytes [27]. Fibroblasts and antigen-presenting cells such as dendritic cells can bind to PtdSer-expressing apoptotic cells via TIM-3 and can mediate their phagocytosis. T cells associated with the TIM-3 receptor are also able to sense PtdSer and interact with apoptotic cells, however, they do not contribute to their clearance [28].
HMGB1 (encoded by the HMGB1 gene) is a plasma protein showing a low cell type specificity as it is expressed in multiple cells and tissues in humans [18]. It is involved in various cellular processes, influences and activates both innate and adaptive immune responses that are mediated by sensors of nucleic acids [29, 30]. HMGB1 is reported to interact with TIM-3 on dendritic cells associated with tumor microenvironments and it is presumed that TIM-3 could negatively regulate innate immune responses promoted by HMGB1 and triggered by nucleic acids. It was further reported that the HMGB1/TIM-3 complex can diminish T cell responses and T cell proliferation [31].
CEACAM1 (encoded by the CEACAM1 gene) is highly expressed in the human gastrointestinal tract on RNA and protein levels followed by the human bone marrow and lymphoid tissues [18]. RNA single cell type specificity identified mainly distal enterocytes and Paneth cells that express CEACAM1 [18]. It appears mainly in context of infectious diseases, autoimmunity and has a tumor-associated function especially in colorectal cancer [32]. It is known as an inhibitory factor on T cells by modulating the activation threshold of T cells [33]. However, it can also be co-expressed together with TIM-3, where it shows tolerance induction of T cells [16]. CEACAM1 can bind to TIM-3 receptors [16]. Subsequently, intracellular processes are stimulated leading to inhibited immune responses that dampen anti-tumor signaling [13].
Role of the TIM-3 receptor in autoimmune and inflammatory bowel diseases
After the structural characterization of TIM-3 and the identification of its ligands, attention is directed to its role in autoimmune diseases, with a focus on IBD. The effects of TIM-3 absence or dysfunction are noteworthy. Genetic alterations of HAVCR2, as reported and identified by Gayden et al. and Polprasert et al. were associated with a TIM-3 deficiency, resulting in uncontrolled immune activation and the increased production of pro-inflammatory cytokines, such as TNF-α. This was attributed to impaired TIM-3 surface expression resulting from protein misfolding [34, 35]. Another study by Zhang et al., investigated the relationship between HAVCR2 polymorphisms and the risk of autoimmune diseases and identified the TIM-3+4259A>C polymorphism to be associated with rheumatoid arthritis [36]. In another study, Baldrich et al. also showed that the absence of TIM-3 directly influences the development of IBD. They delineated the clinical profile of a patient who developed IBD after allogeneic stem cell transplantation. Expression of TIM-3 was minimal on immune cells within the patient’s intestinal mucosa. Whole exome sequencing and Sanger sequencing of the donor-derived DNA unveiled the presence of a rare HAVCR2 missense mutation, denoted cA291G; p.I97M, which resulted in the aberrant folding of TIM-3 and the loss of its surface expression [10]. Overall, the results from genetic modifications of HAVCR2 show that impaired TIM-3 function leads to dysregulated immune activation and increased proinflammatory cytokines, which is also frequently observed in autoimmune diseases.
Furthermore, TIM-3 dysregulations have been implicated in the pathophysiology of IBD. Increased TIM-3 mRNA expression levels were detected by Kim et al. in the colonic mucosa from patients with Crohn’s disease (CD) compared to healthy controls. The same group reported a positive correlation of the severity of inflammation with the expression of HAVCR2, and that elevated TIM-3 expression could be downregulated by the treatment with infliximab [37]. Studying PBMCs and colon tissue samples from patients with ulcerative colitis (UC) or mice with DSS-induced colitis, Shi et al., detected a dysregulation of TIM-3 compared to controls [38]. Contrary to Kim et al. TIM-3 transcript was significantly diminished in colon samples from UC patients and colitis mice. Additionally, enhanced T_H_17 cell function was observed in UC patients and Shi et al., proposed that the increased T_H_17 cell activity might be the consequence of downregulated TIM-3 expression [38]. To explore potential interventions for TIM-3 dysregulation, Shi et al. evaluated the effects of antagonistic α-TIM-3 antibodies and recombinant Gal-9 in murine models of colitis. The Injection of α-TIM-3 antibodies resulted in exacerbated tissue damage and enhanced transcript and protein levels of the proinflammatory cytokines IL-17 and IFN-γ, whereas Foxp3 mRNA levels were alleviated. Moreover, treatment with the TIM-3 ligand Gal-9 improved colitis progression, decreased weight loss and diminished the expression of IL-17 and IFN-γ [38]. The impact of blocking TIM-3 in vivo was also tested by Li et al. in a TNBS-induced colitis model. They detected a significantly impaired disease outcome marked by increased weight loss, mortality, histological evaluated tissue damage and elevated expression of pro-inflammatory cytokines. In further analysis, they reported a decreased percentage and number of CD4^+^Foxp3^+^ Tregs. Surprisingly, blocking TIM-3/Gal-9 signaling did not affect apoptosis of CD4^+^ cells significantly [39]. Shi et al., and Li et al., suggested that the negative impact of theTIM-3 blockade mostly results from enhanced T_H_17 function and attenuated Treg cell response [38, 39]. This goes in line with data from Gautron et al., as they showed that TIM-3^+^ Tregs cells can specifically inhibit T_H_17 cells, an ability that Tregs lacking TIM-3 cannot. Stimulating TIM-3 might enhance the suppressor function of regulatory T cells while blocking results in the opposite effect [40]. A recent study from Xiong et al., focused on the effect of the TIM-3 ligand Gal-9 in induced colitis in mice. Administration of Gal-9 led to overall ameliorated disease severity in TNBS-induced colitis but not DSS-induced colitis. Furthermore, decreased serum levels of IFN-γ, IL1-β, and IL-6 were detected [41]. Overall, the data indicate that TIM-3 blockade promotes IBD severity, whereas TIM-3 activation improves symptoms, with multiple T cell subsets contributing to these TIM-3 mediated effects. Despite these findings, several aspects remain to be critically discussed. While Kim et al. reported increased TIM-3 expression in the colonic mucosa of CD patients, contrary effects were found in isolated PBMCs by the same group and additionally studies from Shi et al. [37, 38]. Ambivalent TIM-3 expression patterns were also described in context with other disorders. T cells from patients with allergic asthma or chronic hepatitis C infection expressed elevated levels of TIM-3, whereas sarcoidosis patients showed downregulated levels of TIM-3 in their lungs [42–44].
In summary, TIM-3 expression and function are inconsistent and can vary depending on the tissue and disease context, contributing to the ongoing debate whether TIM-3 acts as a co-inhibitory or co-stimulatory receptor. A few studies have reported that TIM-3 can promote inflammatory processes, function as a co-stimulatory receptor, and enhance TCR signaling under specific conditions [12, 20, 45, 46]. Lee et al. described an in vitro setting in which murine CD4^+^T cells under Th1 stimulation and infected with retrovirus encoding TIM-3 initially can augment T-cell activation and increase IFN-γ production. However, cross-linking TIM-3 with an agonistic antibody rapidly inhibited T-cell activation [20]. Comparable effects were reported by Avery and colleagues. In their study, enforced TIM-3 expression was accompanied by enhanced TCR signaling in primary T cells and elevated levels of short-lived effector cells [12]. Further in vitro models suggested TIM-3-mediated co-stimulation via MEK-ERK or Akt-mTOR signaling [45] and a recently published study from Alamir et al. described TIM-3 as an inhibitory receptor on spheroid-suppressed cytotoxic T lymphocytes but not on active cells in a two-dimensional tissue culture model. [46]. Taken together, this underlined that TIM-3’s role is ambivalent highly depending on the functional context model applied. On the other hand, findings from HAVCR2 polymorphisms and modulation experiments in autoimmune diseases and IBD, using TIM-3 with blocking antibodies or activation by its ligand galectin-9, rather indicate a predominant inhibitory role, particularly in colitis (Fig. 1) [34, 35, 38, 39, 41].Fig. 1TIM-3 in Autoimmunity Genetic susceptibility in humans and functional evidence from experimental models. HAVCR2 polymorphisms in humans overall increase the susceptibility for diseases such as asthma, rheumatoid arthritis, or IBD (left). In in vivo experimental models, blocking TIM-3 is associated with increased disease severity in COPD, EAE, diabetes, autoimmune hepatitis or IBD. Contrarily, TIM-3 signaling activation can ameliorate disease severity (right) [7, 10, 11, 16, 34, 35, 36, 38, 39, 41, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59]
Intracellular signaling mediated by TIM-3 and the connection to TCR signaling
As TIM-3 can regulate T cell activation, its proximal connection to TCR signaling will be explained in more detail. Intracellular motifs of the TCR need to be phosphorylated by the lymphocyte cell-specific protein-tyrosine kinase (Lck) or on less account by the Src family tyrosine kinase Fyn, in order to activate TCR signaling. Crucial for this phosphorylation step is the location of Lck in proximity to the cell membrane [60]. TIM-3 can mediate the migration of active Lck towards the cell membrane. In the absence of a TIM-3 ligand, the human leukocyte antigen B (HLA-B)-associated transcript 3 (Bat3) is linked to two out of five tyrosine residues (Y265 and Y272) on the intracellular tail of TIM-3 which facilitates Lck recruitment and can contribute to TCR signaling [14, 15, 61, 62]. Contrarily, upon binding of TIM-3’s ligand Galectin-9, Y265 and Y272 tyrosine residues can get phosphorylated either by IL2-inducible tyrosine kinase (ITK) or Src family kinase Lck [15]. As a consequence, Bat3 initiates the inactivation of Lck via the C-terminal Src kinase CsK, that phosphorylates the inhibitory site of Lck [20, 61]. Subsequently, TCR signaling can be interrupted and cell proliferation inhibited due to TIM-3 activation [61]. Simultaneously, stimulation of TIM-3 via Gal-9 can upregulate the expression of CD45 which can facilitate the dephosphorylation of the active form of Lck thereby diminish Lck activity and dampening TCR signaling [63]. Taken together, the TIM-3 receptor contributes to TCR signaling when it is unbound by a ligand and its intracellular tyrosine residues are linked to Bat3. Conversely, TIM-3 can dampen TCR signaling when a ligand is bound and the tyrosine residues of the intracellular tail are phosphorylated by Lck or ITK (Fig. 2) [20]. The ligand-dependent loss of Bat3 from TIM-3 and the ITK-mediated phosphorylation of its intracellular tyrosines together indicate how TIM-3 can influence early steps of TCR signaling.Fig. 2. Proposed schematic interplay of TIM-3 with TCR signaling. Upon stimulation of TIM-3 by Galectin-9 (red pathway), increased CD45 expression leads to dephosphorylation of the activating tyrosine (Y394) on Lck, reducing its kinase activity and suppressing downstream TCR signaling. Concurrently, Gal-9 binding induces phosphorylation of the intracellular tyrosines Y265 and Y272 on TIM-3 and displacement of the inhibitory adaptor Bat3. This process inhibits NFAT/AP-1-driven transcription and promotes T cell exhaustion or apoptosis. ITK may further enhance this phosphorylation, potentially creating a negative feedback loop to attenuate T cell activation. Conversely, in the absence of Gal-9 (green pathway), TIM-3 remains unphosphorylated and retains Bat3, which supports Lck function and TCR signaling. Low CD45 levels enhance Y394 phosphorylation on Lck and maintain its activity, promoting downstream signaling through Zap70 and ITK, ultimately leading to NFAT/AP-1 activation and T cell proliferation. Without TIM-3 activation, the production of IFN-γ and IL-17 cytokines is generally preserved, whereas activation of TIM-3 by Gal-9 suppresses IFN-γ and IL-17 cytokine production, as postulated in experimental models (see also Fig. 1)
Moving to the further downstream events, the TCR signaling cascade involves the binding of zeta-chain-associated protein kinase 70 (Zap-70), a key step that allows Lck to phosphorylate and activate Zap-70. Subsequently, phospholipase C gamma 1 (PLCγ1) gets activated by ITK and the 2nd messengers namely inositol trisphosphate (IP3) and diacylglycerol (DAG) are produced that initiate the translocation of the transcription factors nuclear factor of activated T cells (NFAT), nuclear factor κB (NFκB), and activator protein-1 (AP-1) in the nucleus. Subsequently, cell proliferation- and activation-associated genes get transcribed, which is accompanied by cytokine release [60]. In context with TIM-3, it was reported that TIM-3 can limit the available pools of Lck and PLCγ1 upon stimulation with anti-CD3/anti-CD28 antibodies leading to interrupted TCR signaling [64]. Additionally, Lee et al. demonstrated a dependence of ZAP70 and the adaptor protein SLP-76 for TIM-3-mediated TCR activation [20]. Moreover, point mutations within the intracellular tyrosine residues of the TIM-3 receptor were found to restore NFAT activity [64]. The cytoplasmic tail of TIM-3 is also required for the suppression of both IL-2 production and AP-1/NFAT activation [65], suggesting that these residues are central for the immunoinhibitory signaling of TIM-3. In summary, evidence from several studies suggests that TIM-3 modulates TCR signaling by interacting with it in different steps of the cascade.
Open questions in the TIM-3 signaling pathway: what role does ITK play?
TIM-3 signaling is closely linked to TCR signaling and both pathways are shown to influence each other. As described above, interactions of TIM-3 with Lck, Fyn, ITK, Zap70, or PLCγ1 were reported. Off note, the corresponding genes HAVCR2, Lck*, *Fyn Zap70, or PLCG1 are all located on different chromosomes, except for HAVCR2 and ITK [18]. Both HAVCR2 and ITK are located on chromosome 5, cytoband q33.3, in close proximity (Fig. 3). Interestingly, the gene region on chromosome 5.q33 is also the home of T_H_2 cytokines, which are frequently associated with autoimmune diseases in humans [66]. Not only TIM-3 but also ITK is involved in the development and maintenance of autoimmune diseases, like asthma and multiple sclerosis, or inflammatory bowel disease [10, 36, 38, 41, 67, 68]. ITK can phosphorylate the intracellular tyrosine residues of TIM-3 in HEK293T cells [15]. Phosphorylation of these tyrosine residues reportedly leads to apoptosis of T_H_1 cells and a downregulation of the T_H_1 immune response [15]. Based on the considerations outlined above, the involvement of ITK in TIM-3 signal transduction appears to be of particular interest. While the roles of Lck and Bat3 in TIM-3 signaling are well characterized, the contribution of ITK remains insufficiently understood but might act as an important regulatory node in the TCR signaling network balancing activation and exhaustion. The question arises why ITK, in particular, is capable of phosphorylating the tyrosine residues of the intracellular tail and underscores the need for further investigation.Fig. 3. Genomic location of the genes for TIM-3 and ITK. HAVCR2 and ITK are located on chromosome 5.q33 in humans and chromosome 11B1 in mice respectively. A region known for its association with the development of autoimmune diseases. Abbreviations: Mb (Megabases)
Several questions remain unanswered about the TIM-3/ITK connection in this regard. Notably, among T-helper cell types, T_H_2 cells exhibit the highest levels of ITK, while highest levels of TIM-3 are found on T_H_1 cells [7, 67, 68]. In contrast, T_H_1 cells display lower ITK activity compared to other T_H_ cell subtypes [69, 70]. The phosphorylation of the intracellular tail of TIM-3 by ITK and the subsequent apoptosis could also be the reason for low ITK expression levels in T_H_1 cells, as T_H_1 cells with elevated ITK expression levels are more likely to go into apoptosis. It remains to be clarified in more detail if phosphorylation of the intracellular tail of TIM-3 by ITK in T_H_1 cells induces apoptosis in a direct ITK-dependent manner. A direct interaction between TIM-3 and ITK, or parallel to the interaction between LCK and TCR signal transduction, is an interesting question that cannot be fully answered based on the currently available data. In our opinion, there is a neglected connection between TIM-3 and ITK, or rather an interplay whose investigation could clarify the exact molecular downstream signaling of TIM-3. Further elucidation of these circumstances through more in-depth experiments, particularly in the context of the individual T_H_ cell subtypes, is necessary.
Conclusion
TIM-3 is an important immune checkpoint receptor and a promising target in cancer and autoimmune therapies. The underlying signaling mechanisms remain only partially elucidated, although several studies have demonstrated links to TCR signaling, as summarized in Table 1. Notably, TIM-3 signaling appears to be interconnected with TCR signaling, since TCR-associated kinases such as Lck, Fyn, and ITK can modulate the tyrosine residues of the intracellular tail of TIM-3, conversely, TIM-3 can influence TCR signaling. In context with autoimmunity and inflammatory disorders like IBD, the linkage of TIM-3 and ITK is interesting even though insufficiently investigated. Their close proximity on the same chromosome and their implied direct interaction is a promising connection that should be examined more closely in future research approaches. Table 1. Interactions of TIM-3 with the TCR signaling pathwayInteraction describedExplanationRefLCK → TIM-3TIM-3 → PI3KPhosphorylation of tyrosines 256/263 of TIM-3 at the intracellular tail and recruitment of p85 PI3K is necessary for TIM-3 downstream signaling to NFAT/AP-1 and NF-kB. Src family kinases LCK and FYN can phosphorylate TIM-3. The dependence of src family kinases and ZAP70 and SLP76 proteins in TIM-3 downstream signaling indicates a proximity to the TCR signaling pathway[20]TIM-3 → LCKUpon binding of Gal-9, increased levels of CD45 are detected leading to diminished LCK activity by dephosphorylation of the active form of LCK (pY394) and dampening TCR signaling[63]ITK → TIM-3ITK phosphorylates TIM-3 in HEK293T cells at Y265 of the cytoplasmic tail. Interaction of Gal-9 with TIM-3 leads to Y265 phosphorylation, resulting in impaired TCR signaling[15]TIM-3 → LCK and PLCγ1TIM-3 → NF-kB/NFAT activityTyrosine residues Y265/Y272 at the cytoplasmic tail of TIM-3 are critical for the function. TIM-3 interrupts downstream signals originating from TCR by inhibiting NF-kB/NFAT activation and IL-2 expression. TCR stimulation enables association of Src kinase Lck, and PLCg with TIM-3. TIM-3 inhibits LCK and PLCg, limiting them to carry out the activation steps required for TCR signaling[64]TIM-3 → PI3KTCR stimulation with αCD3/CD28 as well as γc cytokines IL-2, IL-7- IL-15 and IL-21 drives TIM-3 expression on CD4^+^/CD8^+^ T cells in vitro. TIM-3^+^ T cells are more sensitive to apoptosis through galectin-9. Blocking the PI3K pathway by using a PI3K inhibitor diminished TIM-3 expression[71]TIM-3 → NFAT/AP-1TIM-3 expression reduced the stimulation-induced dephosphorylation and nuclear translocation of NFAT. The cytoplasmic tail was necessary for IL-2 suppression as well as AP-1 and NFAT activation. T cell IL-2 production may be suppressed by TIM-3, leading to reduction of NFAT dephosphorylation and AP-1 transcription[65]
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Huang Y-H et al (2015) CEACAM 1 regulates TIM-3-mediated tolerance and exhaustion. Nature 517(7534): p. 386–390. corrigendum: 10.1038/nature 1742110.1038/nature 13848 PMC 429751925363763 · doi ↗ · pubmed ↗
- 2Consortium TU (2022) Uni Prot: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res 51(D 1): p. D 523–D 531.10.1093/nar/gkac 1052 PMC 982551436408920 · doi ↗ · pubmed ↗
- 3Lv Y, Ma X, Ma Y, Du Y, Feng J. (2022) A new emerging target in cancer immunotherapy: Galectin-9 (LGALS 9). Genes Dis 10(6):2366-2382. 10.1016/j.gendis.2022.05.02010.1016/j.gendis.2022.05.020PMC 1040487737554219 · doi ↗ · pubmed ↗