Convergent subgenome dominance but with lineage-specific functional divergence of homoeologs during cave adaptation: insights from full-length transcriptomes of Sinocyclocheilus species
Shaohua Xu, Mingming Zhang, Fanwei Meng, Chongnv Wang, Xinxin Li, Baocheng Guo

TL;DR
This study explores how duplicated genomes in cavefish evolved to help them adapt to cave environments, showing that subgenome dominance leads to different adaptations in different species.
Contribution
The study reveals convergent subgenome dominance and lineage-specific functional divergence in cavefish, linking genome duplication to ecological adaptation.
Findings
Cavefish species show B-subgenome dominance in homoeolog expression, while surface species show balanced expression.
Subgenome-dominant genes in S. microphthalmus are enriched in immunological elements, indicating immune investment shifts.
Subgenome-dominant genes in S. furcodorsalis are enriched in neuromodulatory and metabolic pathways, suggesting energy conservation.
Abstract
Allopolyploidy creates duplicated genomes that drives evolutionary innovation and adaptive diversification under extreme environmental pressures. Although subgenomic architecture is recognized as pivotal in post-polyploid evolution, the mechanisms by which divergent subgenome dynamics shape adaptive potential remain unclear. We investigated how subgenome evolution relates to environmental adaptation in Sinocyclocheilus cavefish, an allotetraploid lineage that repeatedly colonized caves across the karst landscapes of Southwest China. We integrated full-length and short-read transcriptomes from a surface-dwelling species (S. angustiporus) and two independently cave-adapted species (S. microphthalmus and S. furcodorsalis). The two cave dweller species showed consistent B-subgenome dominance in homoeolog expression, whereas the surface species showed balanced expression. Functional…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
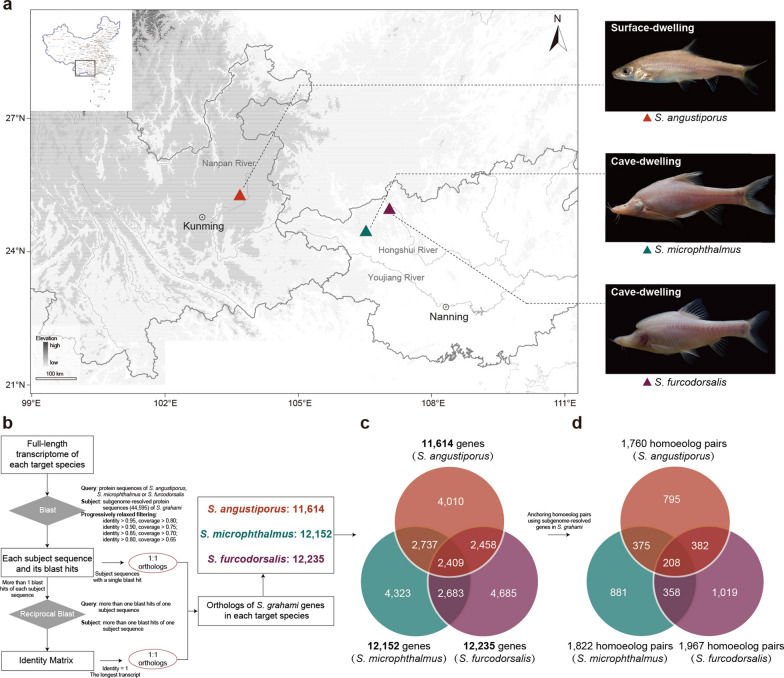 Figure 1
Figure 1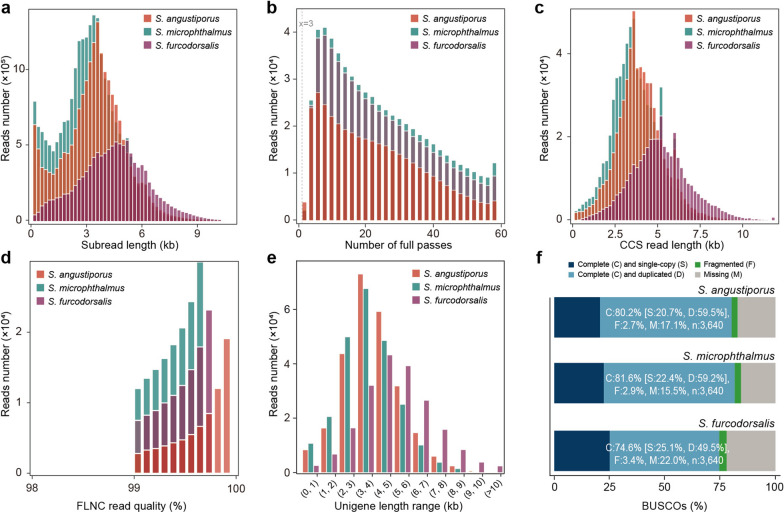 Figure 2
Figure 2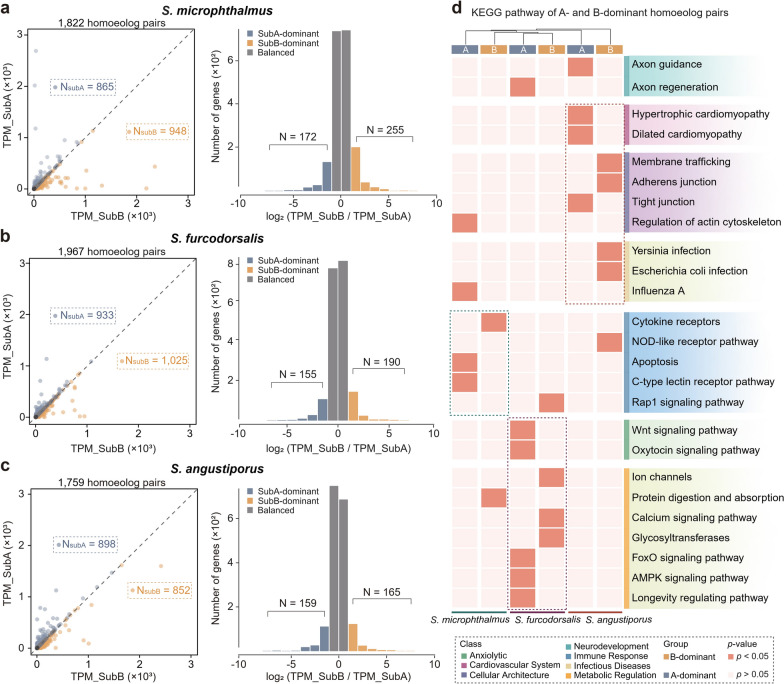 Figure 3
Figure 3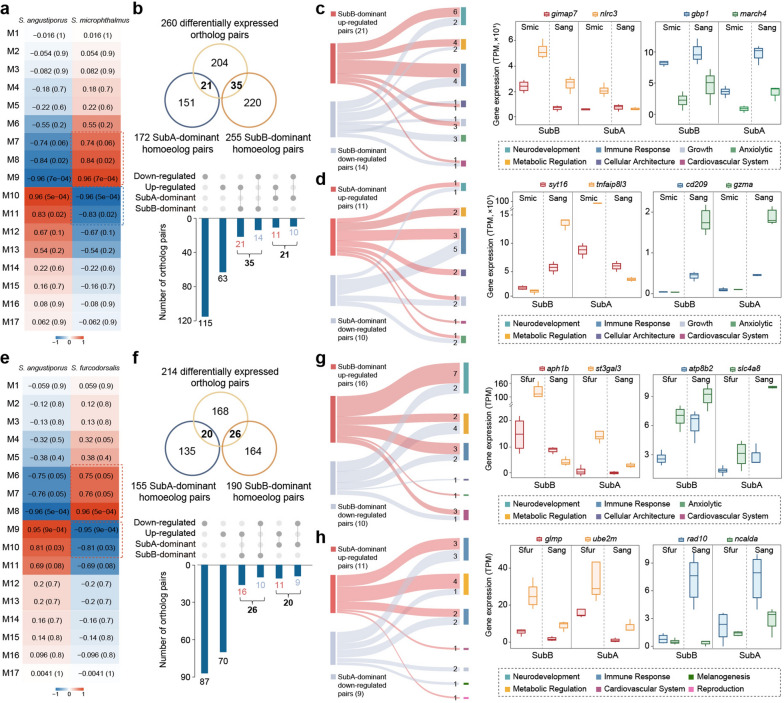 Figure 4
Figure 4- —The National Natural Science Foundation of China
- —The Ministry of Science and Technology of the People’s Republic of China
- —Supercomputing Center in Yancheng
- —Institute of Zoology, Chinese Academy of Sciences
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSubterranean biodiversity and taxonomy · Chromosomal and Genetic Variations · Genomics and Phylogenetic Studies
Introduction
Polyploidization constitutes a major evolutionary force, generating genomic plasticity that enhances adaptive potential under extreme environmental conditions [1–3]. While recurrent polyploidy is widespread among angiosperms, the mechanisms by which genome duplication drives environmental specialization, particularly in harsh and isolated ecosystems, remain a fundamental question in evolutionary genomics [1]. Cave environments, characterized by perpetual darkness and nutrient scarcity, provide powerful natural laboratories for exploring how polyploid genomic architectures influence adaptive trajectories through subgenome evolution [4]. Cave-adapted organisms, spanning a broad taxonomic range from invertebrates to mammals, have emerged as critical models for dissecting the genomic basis of adaptation to extreme environments [5–7].
The cyprinid genus Sinocyclocheilus, comprising 83 recognized species [8–13], represents the most species-rich radiation of freshwater cavefish known to date [14]. Members of this genus exhibit remarkable morphological diversity, spanning surface-dwelling forms with fully developed eyes and pigmentation to cave-adapted phenotypes characterized by eye and scale degeneration, reduced or absent pigmentation, and extensive sensory system remodeling [15–19]. Distinct from most cavefish models, such as Astyanax mexicanus, Sinocyclocheilus underwent a recent shared allotetraploidization event with common carp and goldfish [20, 21], resulting in a composite genome formed from two divergent cyprinid progenitors before colonization of the karstic landscapes of Southwest China [22]. This allotetraploid architecture establishes Sinocyclocheilus as a unique vertebrate system for exploring the evolutionary consequences of genome duplication in extreme environments [23]. Its taxonomic richness, diverse phenotypic adaptations, and duplicated genome structure make the genus an excellent multi-species model for investigating the functional interplay between polyploidy and extreme ecological specialization [18, 24, 25].
To elucidate the regulatory architecture underlying adaptation to extreme subterranean environments, a subgenome-resolved transcriptomic comparison was conducted across three Sinocyclocheilus species, including the surface-dwelling S. angustiporus and the cave-dwelling S. microphthalmus and S. furcodorsalis, which represent phylogenetically divergent lineages exhibiting convergent adaptation to cave environments [8]. Full-length transcriptomes generated via PacBio Iso-Seq and Illumina short-read sequencing enabled the construction of high-confidence subgenome-resolved homoeolog annotations and quantitative expression profiles. Analysis revealed consistent B-subgenome dominance (SubB-dominance) in the phylogenetically distinct cave-dwelling species, in sharp contrast to the balanced subgenome dynamics observed in the surface relative. This convergent regulatory bias was associated with lineage-specific functional divergence, characterized by the subgenomic repurposing of immune-related modules in S. microphthalmus and neuromodulatory and metabolic pathways in S. furcodorsalis. These findings suggest that polyploidy drives adaptation to cave environments through convergent subgenome dominance. In this process, selective regulatory asymmetry resolves ancestral genomic conflicts by channeling stress-responsive pathways into lineage-specific functional trajectories. This mechanism establishes a link between genome duplication and ecological innovation.
Materials and methods
Sampling, sequencing, assembly, and annotation
Specimens of the three Sinocyclocheilus species were collected from Yunnan Province and Guangxi Zhuang Autonomous Region (Fig. 1a). For each species, three adult individuals were sampled. Brain, liver, muscle, and skin tissues were dissected and immediately frozen in liquid nitrogen for sequencing. The full-length transcriptomes of four mixed tissues were sequenced using the PacBio Sequel II platform. Additionally, brain-specific transcriptomes were sequenced using the DNBSEQ-T7RS platform (Nextomics Biosciences Co., Ltd., Wuhan, China). All procedures involving animals were approved by the Animal Care and Use Committee of the Institute of Zoology, Chinese Academy of Sciences (approval number: IOZ18002).Fig. 1. Sampling information and homoeolog pair identification based on full-length transcriptomes of three Sinocyclocheilus species. a Sampling information of three Sinocyclocheilus species in this study. b Identification pipeline of S. grahami orthologous genes in each species. c Venn diagram showing unique and shared orthologs (1:1:1) among the three species*.* d Venn diagram showing unique and shared homoeolog pairs (1:1:1) among the three species
Raw subreads from the PacBio platform were processed using the Iso-Seq3 pipeline (https://github.com/PacificBiosciences/IsoSeq3) to assemble high-quality full-length transcriptomes. Circular consensus sequences (CCS) were generated using ccs v6.4.0 with default parameters. Full-length (FL) and non-full-length (NFL) reads were identified based on the presence or absence of 3′ and 5′ primers and a Poly(A) tail. Primer removal and poly(A) tail trimming were performed using Lima v2.9.0 and Refine v4.0.0, respectively, with default parameters. Finally, the full length non-chimeric reads (FLNC) sequences were clustered using cluster v4.0.0, followed by extraction of high-quality isoforms and redundancy removal using cd-hit v4.8.1 (identity > 99%). Transcriptome completeness after de-redundancy was assessed using BUSCO v5.3.0 [26].
Non-redundant transcript sequences (unigenes) were subjected to open reading frame (ORF) prediction using the ANGEL tool (https://github.com/PacificBiosciences/ANGEL) with default parameters. Coding (CDS) and corresponding amino acid sequences were extracted for functional analysis. Gene annotation was performed using BLAST v2.12 [27], with a significance cutoff of E-value < 1E−5 and other parameters set to default. Functional classification of unigenes was conducted using Gene Ontology (GO) [28] and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases [29]. Transcription factors (TFs) were identified using AnimalTFDB v4.0 (https://guolab.wchscu.cn/AnimalTFDB4) and classified into TF gene families [30] using HMMER v3.0 [31]. Simple sequence repeats (SSRs) were detected using the MISA tool (http://pgrc.ipk-gatersleben.de/misa/), which categorizes microsatellite motifs into mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide classes.
Identification of orthologs and homoeolog pairs
To resolve subgenomic structure, the S. grahami genome [18] was partitioned into A and B subgenomes using Cyprinus carpio as a reference, based on BLASTN alignments [22]. BLAST v2.2.26 [27] was then used to align the predicted peptide sequences from the three Sinocyclocheilus species against the above subgenome-resolved protein sequences of S. grahami with an E-value threshold of 1E−3 (Fig. 1b). Hits were filtered using stringent criteria (“identity > 0.95 and coverage > 0.8”) to identify putative orthologs (Fig. 1b). Transcripts without BLAST hits were classified as 1:0 orthologs, while those with a single high-confidence hit were categorized as 1:1 orthologs (Fig. 1b). In cases where multiple hits were detected, the transcript showing the highest sequence identity and full-length alignment was retained as the best representative 1:1 ortholog. To increase ortholog recovery, the mapping results were further filtered using three progressively relaxed parameter sets: “identity > 0.9 and coverage > 0.75”; “identity > 0.85 and coverage > 0.7”; “identity > 0.8 and coverage > 0.65” (Fig. 1b). Orthologs identified under these relaxed thresholds were merged iteratively to expand the ortholog catalog between each species and the reference species (Fig. 1b). From these results, 1:1:1 orthologs shared among all three Sinocyclocheilus species were extracted (Fig. 1c). Homoeolog pairs specific to each species were subsequently identified from this ortholog set, providing a high-confidence set of 1:1 homoeologs for downstream subgenome expression analyses (Fig. 1d).
Weighted gene co-expression network analysis (WGCNA)
High-quality transcriptome reads were generated by trimming raw sequencing data with fastp v0.12.4 [32] and alignment to species-specific CDS using BWA v0.7.17-r1188 [33]. Gene expression was quantified using Salmon v1.10.2 [34], with expression levels normalized to transcripts per million (TPM). Orthologous and homoeologous gene sets (Fig. 1c, d) were used to extract matched expression profiles across the three Sinocyclocheilus species. For each homoeologous gene pair, the copy exhibiting at least a twofold higher expression [20, 35] than the other was considered to have dominant expression. A two-sided chi-squared test, as described in previous studies [22, 35–37], was performed to test whether the dominant gene number in the A subgenome significantly differed from that in the B subgenome. WGCNA was performed using the R package WGCNA v1.72-1 [38], focusing on genes with non-zero TPM values in brain tissue. Co-expression networks were constructed independently for comparisons between S. angustiporus and S. microphthalmus and between S. angustiporus and S. furcodorsalis, to identify species-specific gene modules associated with cave adaptation. Modules exhibiting strong positive or negative correlations (correlation coefficients ≥ 0.70 or ≤ − 0.70) with statistically significant associations (p < 0.05) were retained for further analysis. Within each module, gene significance (GS) and module membership (MM) were calculated, and core module genes were defined by GS > 0.40 and MM > 0.50.
Functional enrichment analysis
KEGG pathway enrichment analysis of differentially expressed genes (p < 0.05) was performed using the clusterProfiler v4.4.4 R package [39]. The top 10–15 enriched pathways were selected and visualized using the ggplot2 v3.5.1 R package [40].
Results
High-quality, full-length transcriptomes of three Sinocyclocheilus species
High-fidelity PacBio Iso-Seq data were generated for the three Sinocyclocheilus species to construct high-resolution transcriptomic landscapes. Sequencing yielded 20,074,273, 23,654,573, and 11,589,089 subreads, with average lengths of 3.54, 3.23, and 4.63 kb and N50 lengths of 4.15, 3.87, and 5.26 kb, respectively (Fig. 2a and Table S1). Subsequent processing of subreads with more than three full passes produced 620,461, 683,050, and 401,523 CCS reads (Fig. 2b), with average lengths of 3.99, 3.71, and 5.21 kb and N50 lengths of 4.33, 4.12, and 5.64 kb, respectively (Fig. 2c and Table S1). FLNC transcripts were identified through poly(A) tail and primer detection, resulting in 613,077 (S. angustiporus), 676,574 (S. microphthalmus), and 398,596 (S. furcodorsalis) high-confidence transcripts (Table S1). After clustering and redundancy reduction, 256,272, 237,700, and 196,497 non-redundant unigenes were retained, each exhibiting sequence identity larger than 99% (Fig. 2d and Table S1). Length distribution analysis revealed that most unigenes exceeded 2 kb—90.41% in S. angustiporus, 86.89% in S. microphthalmus, and 95.32% in S. furcodorsalis (Fig. 2e and Table S3). BUSCO assessments confirmed transcriptome completeness, detecting 80.2% (single-copy: 20.7%, duplicated: 59.5%), 74.6% (single-copy: 22.4%, duplicated: 59.2%), and 81.6% (single-copy: 25.1%, duplicated: 49.5%) complete orthologs, respectively, suggesting high-quality full-length transcriptomes (Fig. 1f).Fig. 2. Quality evaluation for reads generated using the Iso-seq3 analysis pipeline. a Length distribution of subreads. b Length distribution of subreads with more than three full passes. c Length distribution of CCS reads. d Quality distribution of full length non-chimeric reads (FLNC) reads. e Length range distribution of unigenes. f BUSCO completeness metrics for full-length transcriptomes
Coding sequence (CDS) prediction yielded 233,965 (S. angustiporus), 212,345 (S. microphthalmus), and 190,606 (S. furcodorsalis) CDSs in the three species, with length distributions of 90.97%–94.87% below 3.0 kb, 4.67–7.42% between 3.0 and 5.0 kb, and 0.46–1.61% exceeding 5.0 kb (Table S3). Notably, over 95% of CDSs encoded more than 1,200 amino acids across all species (Fig. S1 and Table S3). Analysis of TFs using AnimalTFDB v4.0 identified 57,548 TFs in S. angustiporus, 51,702 TFs in S. furcodorsalis, and 52,719 TFs in S. microphthalmus, distributed across 72 gene families (Fig. S2a and Table S4). In parallel, SSR profiling revealed a strong bias toward shorter repeat motifs, with mononucleotide and dinucleotide repeats accounting for over 60% and 25% of SSRs across all three species (Fig. S2b and Table S5).
Convergent B-subgenome expression dominance in two cave-dwelling species
To investigate subgenome regulatory asymmetry associated with cave adaptation, transcriptional profiling was conducted on 1,822, 1,967, and 1,759 high-confidence homoeolog pairs in S. microphthalmus, S. furcodorsalis, and S. angustiporus, respectively (Fig. 3a–c left). Comparative transcriptomic analyses revealed a consistent pattern of expression bias favoring the B subgenome in cave-adapted Sinocyclocheilus species, in contrast to the more balanced subgenome activity observed in their surface-dwelling counterpart. In S. microphthalmus, 948 homoeolog pairs showed elevated expression from the B subgenome relative to 865 from the A subgenome, while S. furcodorsalis exhibited a similar skew with 1,025 vs. 933 gene pairs, respectively (Fig. 3a–b, left). In comparison, S. angustiporus showed a slight shift toward A-subgenome expression (898 vs. 852 gene pairs), indicating no pronounced subgenomic bias (Fig. 3c, left). To delineate dominant expression patterns, homoeolog pairs with at least a two-fold expression bias toward one subgenome relative to its counterpart were classified as dominantly expressed, following established definitions [41, 42]. Notably, S. microphthalmus exhibited significant B-subgenome dominance, with 255 B-dominant versus 172 A-dominant homoeologs (χ^2^ = 16.1335, p = 5.9e-5) (Fig. 3a right and Table 1), while S. furcodorsalis showed a similar directional trend (190 vs. 155), although it did not reach statistical significance (χ^2^ = 3.5507, p = 0.0595) (Fig. 3b right and Table 1). In contrast, S. angustiporus displayed no significant subgenome dominance, with 165 B-dominant and 159 A-dominant homoeolog pairs (χ^2^ = 0.1111, p = 0.7389) (Fig. 3c right and Table 1). This convergent bias toward B-subgenome expression in both cave-adapted species, despite their phylogenetic divergence, suggests that subgenome dominance may represent a key regulatory mechanism. This mechanism may facilitate adaptation to the extreme conditions of subterranean environments. Notably, to rule out effects of the cutoff choice, we also tested a 1.5-fold cutoff. It identified more genes, but the functional enrichments matched those at twofold. We therefore used the commonly adopted twofold threshold (Fig. S3).Fig. 3. Expression divergence of homoeolog pairs across subgenomes in S. angustiporus, S. microphthalmus, and S. furcodorsalis. a–c Distribution of TPM_SubA/TPM_SubB ratios (left) and expression divergence of homoeologous genes (right) in brain tissue across three species. Dashed line (TPM_SubB/TPM_SubA = 1) indicates equal expression. Blue dots represent homoeolog pairs with a ratio > 1, orange dots denote those with reversed expression bias. Log_2_ (TPM_SubB/TPM_SubA) indicates degree of expression difference in homoeolog pairs. N values indicate number of homoeolog pairs with two-fold expression divergence. d KEGG enrichment analysis of SubA- and SubB-dominant homoeologs in two subgenomes across three species. Pathways are categorized into seven functional groups. Color gradient represents significance level of enrichment, with red indicating p < 0.05Table 1Two-sided chi-squared test for dominant genes expressed in the brain tissue of each speciesSpeciesObserved SubAObserved SubBExpected SubAExpected SubBχ^2^dfp-valueS. microphthalmus172.0255.0213.5213.516.133515.9e−5S. furcodorsalis155.0190.0172.5172.53.550710.0595S. angustiporus159.0165.0162.0162.00.111110.7389
KEGG and GO functional enrichment analyses revealed striking contrasts in the biological roles of dominantly expressed homoeolog pairs across subgenomes between surface- and cave-dwelling species (Figs. 3d and S5). In the surface-dwelling S. angustiporus, dominant homoeolog pairs from both subgenomes were primarily enriched in cardiovascular function (e.g., hypertrophic cardiomyopathy and heart development), immune responses (e.g., Yersinia infection and defense response to virus), and molecular transport pathways (e.g., membrane trafficking), consistent with physiological demands imposed by open-water habitats and sustained mobility [43] (Figs. 3d and S5). In contrast, both cave-adapted species exhibited subgenome specialization aligned with subterranean ecological constraints. Dominant homoeolog pairs in the two species were significantly enriched in immune regulation, including lineage-specific activation of the T cell receptor signaling pathway and inflammatory response in S. microphthalmus and the Rap1 signaling pathway in S. furcodorsalis. Metabolic adaptations were also prominent, with overrepresented pathways such as protein digestion and absorption in S. microphthalmus and MAPK signaling in S. furcodorsalis, hallmarks of survival under nutrient-poor conditions [4, 44] (Fig. 3d). Comparative analyses further revealed species-specific divergence in the functional deployment of dominant homoeolog pairs. In S. microphthalmus, dominant homoeologs from the two subgenomes were primarily involved in immunological modulation. In S. furcodorsalis, however, dominant homoeologs were significantly enriched in anxiolytic and metabolic regulation, mainly via Wnt and MAPK signaling, pathways implicated in behavioral modulation and efficient energy utilization under metabolic constraint (Fig. 3d) [45]. These distinct subgenomic signatures reflect lineage-specific solutions to the physiological demands of cave life and underscore the role of subgenome partitioning in facilitating adaptive divergence.
Together, these results connect genome duplication to ecological adaptation in a testable way. Preferential use of the B subgenome seems to help these fishes cope with cave pressures (e.g., long-term food shortage, perpetual darkness, and shifts in pathogen exposure) by raising adaptive-immune activity and shifting metabolism toward more energy-efficient modes. In this way, the polyploid genome is not a fixed gene store but a modular system with built-in regulatory bias that can be selectively used during cave adaptation in Sinocyclocheilus.
Distinct functional specialization of subgenome-dominant homoeologs
To investigate the evolutionary implications of subgenome dominance in cave-adapted Sinocyclocheilus lineages, subgenome-resolved co-expression networks were constructed by comparing S. angustiporus with the cave-dwelling S. microphthalmus and S. furcodorsalis (Fig. 4a, e and S4a, e). In S. microphthalmus, three modules (M7–9) exhibited strong positive correlation with cave phenotype (r > 0.70, p ≤ 0.05), containing 900 up-regulated genes, while two modules (M10–11) were negatively associated (r < − 0.70, p ≤ 0.05), encompassing 2,095 down-regulated genes. Gene filtering with MM (cor.MM > 0.50) and GS (cor.GS > 0.40) resolved 839 up-regulated and 1 757 down-regulated candidate genes (Figs. 4a and S4b, c). A parallel regulatory structure was observed in S. furcodorsalis, with three positively correlated (M6–8) and two negatively correlated modules (M9 and M10), corresponding to 703 up-regulated and 1,803 down-regulated genes (Figs. 4e and S4e, f). Cross-species comparison of these networks revealed conserved patterns of subgenome-biased expression among differentially expressed homoeolog pairs (Fig. 4b, f). Intersection with cross-species ortholog pairs (Fig. 1b–d) identified 260 and 214 differentially expressed gene pairs in S. microphthalmus and S. furcodorsalis, respectively (Fig. 4b, f). In S. microphthalmus, B-subgenome dominance (SubB-dominant) was evident in 35 gene pairs (21 up-regulated and 14 down-regulated), while A-subgenome dominance (SubA-dominant) was observed in 21 gene pairs (11 up-regulated and 10 down-regulated) (Fig. 4b). Similarly, S. furcodorsalis exhibited 26 SubB-dominant pairs (16 up-regulated and 10 down-regulated) and 20 SubA-dominant pairs (11 up-regulated and nine down-regulated) (Fig. 4f). The convergent transcriptional asymmetry toward B-subgenome activity across both cave-dwelling species highlights a recurrent regulatory signature potentially underpinning lineage-specific adaptation to subterranean environments.Fig. 4. Differentially expressed homoeolog pairs exhibiting subgenome dominance in two cave-dwelling species. a, e Heatmaps showing modules from WGCNA comparing S. angustiporus with S. microphthalmus and S. angustiporus with S. furcodorsalis. Correlation coefficients |r|> 0.7 with p < 0.05 were considered significant. Red and blue dashed boxes indicate up- and down-regulated correlations, respectively. b, f Overlap of differentially expressed ortholog pairs (up- and down-regulated gene pairs) and subgenome-dominant homoeolog pairs (SubA and SubB) in S. microphthalmus and S. furcodorsalis, respectively. c, d, g, h Functional classification of SubA- and SubB-dominant homoeolog pairs (up-regulated in red and down-regulated in blue) and subgenome expression divergence of representative key genes in S. microphthalmus and S. furcodorsalis. Sang, S. angustiporus; Smic, S. microphthalmus; Sfur, S. furcodorsalis. SubA, subgenome A; SubB, subgenome B
To further elucidate the functional relevance of subgenome dominance in cave adaptation, ortholog pairs showing both lineage-specific differential expression and subgenome-biased expression in cave-dwelling species were subjected to functional classification (Fig. 4c, d, g, h, and Table S6). In S. microphthalmus, SubB-dominant gene pairs associated with cave-specific expression shifts were significantly enriched in immune-related pathways, particularly those linked to adaptive immune regulation. Among these, six up-regulated and four down-regulated orthologous gene pairs were identified, with notable involvement in immune effector functions (Fig. 4c). Notably, the two up-regulated pairs of gimap7 (modulating apoptotic balance) [46] and nlrc3 (T-cell response regulation) [47] were associated with adaptive cellular immune responses, while the two down-regulated pairs of gbp1 (inflammasome activation) [48] and march4 (antiviral innate immune signaling pathways) [49] were associated with innate immunity components (Fig. 4c). A-subgenome-dominant gene pairs in S. microphthalmus displayed a complementary immune profile. Up-regulated genes, such as syt16 (B and T cell infiltration) [50] and tnfaip8l3 (proliferation, inflammation, and cell death), indicated enhanced adaptive immunity [51], while down-regulated genes, including cd209 (pathogen recognition receptor) [52] and gzma (cell death during phagocytosis), pointed to attenuated innate immune responsiveness [53] (Fig. 4d). These patterns reveal a subgenome-dependent reorganization of immune investment strategies, with differential emphasis on adaptive versus innate immunity components. The results suggest that subgenome-biased expression is intricately linked to functional specialization in cave environments, where altered pathogen exposure and metabolic constraints may have reshaped immune priorities.
In S. furcodorsalis, subgenome-biased expression revealed functional specialization toward neurodevelopmental delay and metabolic adaptation, consistent with selective pressures in nutrient-deprived, lightless cave environments. Analysis of B-subgenome-dominant orthologs identified seven up-regulated gene pairs enriched in neurodevelopmental pathways. Notably, aph1b (neurodevelopmental disorders) [54] and st3gal3 (intellectual disability and behavioral disorders) [55] were implicated in disrupted synaptic maturation. This transcriptional signature corresponds with established observations of reduced visual system development in cave-dwelling fish species [56]. In parallel, four down-regulated B-dominant orthologs, including atp8b2 (ATP-dependent phospholipid/metal ion transporter) [57] and slc9a7 (Na + /H + homeostasis maintenance) [58], were linked to metabolic regulation. This pattern may potentially reflect physiological adjustments to the chronic energy scarcity typical of subterranean habitats [59] (Fig. 4g). Functional profiling of A-subgenome-dominant orthologs revealed complementary patterns. Four up-regulated orthologs were primarily associated with lipid metabolism, including glmp (fatty acid uptake and lipogenesis) [60] and ube2m (lipid accumulation and obesity phenotypes) [61]. In contrast, three down-regulated A-dominant orthologs were functionally linked to neural development and intracellular transport, such as rad10 (vesicle trafficking mediator) [62] and ncalda (regulator of neurogenesis) [63], reinforcing the transcriptomic signature of attenuated neurodevelopment observed in B-subgenome expression (Fig. 4h). Comparative analysis with surface-dwelling species further revealed divergent subgenome-biased expression in cavefish lineages, shaped by cave-specific environmental pressures (e.g., light deprivation and nutrient scarcity), ultimately driving species-specific adaptations in immune regulation and metabolic reprogramming.
Together, these results strengthen the link between genome duplication and ecological adaptation. The key signals concentrate in WGD-derived homoeologs, indicating that duplication provides the regulatory space for functional reassignment under cave constraints. Moreover, the repeatable B-subgenome bias in two independently cave-adapted lineages points to a shared regulatory solution. Functionally, duplicated copies are differentially deployed: B-linked homoeologs elevate adaptive-immune and stress-response programs, while A-linked sets redistribute metabolism toward more energy-efficient modes under long-term food shortage and shifting pathogen exposure. Thus, WGD supplies the substrate for subgenome-level partitioning that translates molecular bias into cave-relevant physiological adaptation.
Discussion
Comparative subgenome-resolved transcriptomic analysis revealed convergent B-subgenome dominance and lineage-specific functional divergence in cave-adapted Sinocyclocheilus species. This pronounced regulatory asymmetry underscores subgenome dominance as a key mechanism facilitating adaptation to the extreme conditions of subterranean ecosystems. It also provides novel insights into how polyploid genomic architectures mediate evolutionary innovation under environmental stress.
The consistent expression bias toward the B subgenome across independently evolved cave lineages mirrors trends in polyploid relatives such as common carp [20, 35] and goldfish [22]. Common carp and goldfish share the Cyprinidae-specific whole-genome duplication (Cs4R) event with Sinocyclocheilus [20, 22], and their A/B subgenomes are macro-syntenically orthologous across Cyprinidae. In these species, expression is biased toward the B subgenome, with B-dominant homoeologs enriched for hypoxia/stress response, oxidoreductase activity, hydrolase activity, and DNA repair [20, 22]. A similar pattern of subgenome dominance is also observed in cave Sinocyclocheilus species at the chromosome level, where the D subgenome (B subgenome in this study) exhibits gene-retention and expression dominance corresponding to adaptive functions [64]. Consistent with this framework, population-genomic analyses in cave-dwelling S. microphthalmus also show significantly higher SNP density in the B(H) subgenome at the level of syntenic blocks and homoeologs [65]. This pattern is consistent with our observation of recurrent B-subgenome bias in cave Sinocyclocheilus and supports the view that the B subgenome serves as a preferential regulatory reservoir recruited under cave-specific selection. However, it contrasts with the balanced subgenome activity in surface-dwelling S. angustiporus. This pattern suggests that the B subgenome may be a preferential regulatory reservoir recruited under cave-specific selection. The asymmetric expression likely reflects ancestral genomic incompatibilities from the allopolyploid origin of Sinocyclocheilus [20, 21], in which retention of stress-responsive loci is biased toward one parental subgenome. Outside vertebrates, allopolyploid plants adapted to harsh environments show similar subgenome-level functional specialization. Similarly, enrichment of immune and metabolic regulators in B-subgenome-dominant genes resembles findings in polyploid extremophiles such as Trifolium repens [66] and Sesbania cannabina [67]. In these species, subgenome-specific expression of stress response loci (e.g., RafS and AT1) supports tolerance to cold and saline-alkaline soils. Likewise, the xerophytic allopolyploid Lespedeza potaninii shows consistent B-subgenome dominance across multiple tissues, with functional enrichment in carbohydrate and phenylpropanoid metabolism linked to survival under arid, nutrient-limited conditions [68]. These cross-taxonomic parallels suggest that subgenome-biased regulation can channel adaptive shifts in energy allocation, stress response, and tissue remodeling, key processes also implicated in cave adaptation.
The two subgenomes show lineage-specific functional specialization: adaptive immune regulation in S. microphthalmus and metabolic reprogramming in S. furcodorsalis. This highlights the versatility of polyploid genomes in addressing distinct ecological challenges. In S. microphthalmus, immune-regulatory genes from both subgenomes are elevated, including gimap7, nlrc3, syt16, and tnfaip8l3 (Fig. 4c–d). This pattern suggests a prioritization of adaptive immunity over metabolically expensive innate defenses. At the sequence level, the patterns parallel the expression shift. In S. microphthalmus, the immune gene fcgbp shows a lower density of nonsynonymous SNPs in the H (B) copy, whereas the nervous-system genes cacna2d2, pcdha3, and pcdhac2 exhibit higher nonsynonymous SNP densities in the L (A) copy [65]. The dominant copy tends to be more highly expressed and under stronger purifying selection (fewer nonsynonymous changes), these patterns imply B-subgenome expression dominance for fcgbp and A-subgenome dominance for cacna2d2, pcdha3, and pcdhac2. This shift likely reflects trade-offs imposed by open aquatic habitats, where pathogen exposure is higher, requiring efficient immune surveillance that minimizes energetic cost. The semi-open cave habitats of S. microphthalmus are connected to underground rivers linked with surface waters [69, 70]. These habitats may have relatively high pathogen diversity compared to fully enclosed caves, leading to increased pathogen exposure. Innate immunity, however, is energetically costly and can lead to chronic inflammation [71–73], whereas adaptive immunity offers a more efficient long-term solution by relying on memory cells to recognize and respond to pathogens more effectively with less energy consumption [74, 75]. This is particularly advantageous in nutrient-poor cave environment for cavefish. Similar immune remodelling has been reported in other cavefish, such as A. mexicanus and Triplophysa rosa, where T-cell-based immune strategies dominate [76, 77]. The observed immune allocation aligns with the immunocompetence handicap hypothesis [78], wherein immune function is selectively modulated to balance survival and energy efficiency under resource limitations. In contrast, S. furcodorsalis exhibits metabolic specialization optimized for trophically constrained environments. Down-regulation of ion-transport regulators such as atp8b2 and slc9a7 (Fig. 4f) suggests suppression of energetically costly ATP-dependent processes, a strategy observed in hibernating mammals during extended fasting [79]. Concurrent up-regulation of lipid metabolic genes from the A subgenome, including glmp and ube2m (Fig. 4g–h), supports enhanced lipid storage capacity. This metabolic economy is further reinforced by the down-regulation of neurodevelopmental genes such as aph1b and st3gal3, consistent with reduced sensory investment, a well-established feature of cave adaptation [56, 80]. These traits parallel phenotypic trends in A. mexicanus, including reduced sociality [81], aggressiveness [82], and sleep [83], which collectively reduce energy expenditure under persistent resource scarcity. Consistent with these molecular patterns, S. furcodorsalis has no eyes and has far fewer neuromasts, reflecting adaptation to the perpetual darkness in caves [84]. In addition, it also has a dorsal humpback that lacks bone and is mainly composed of adipose tissue [85]. These traits suggest an adaptive energy reallocation strategy, whereby resources are moved away from vision and sensory systems toward long-term fat storage, helping the fish survive in caves where food is persistently scarce. At the genome scale, these functional shifts align with subgenome-level regulatory asymmetry observed in cave lineages [64], suggesting that adaptive immune and metabolic remodeling can be controlled through the dominant subgenome.
Collectively, these findings establish Sinocyclocheilus as a model system for understanding how polyploid genomes resolve evolutionary trade-offs during extreme environmental constraints. The consistent dominance of subgenome B in cave-dwelling species highlights the role of ancestral polyploid subgenomes as reservoirs of adaptive capacity, selectively deployed to meet ecological challenges. These findings contribute to the growing intersection of polyploid genomics and extremophile research, offering a framework for investigating the genetic basis of resilience in rapidly shifting environments. Nonetheless, key questions remain. First, the role of post-polyploidization diploidization, such as fractionation bias and chromosomal rearrangements, in shaping subgenome architecture remains unclear. Second, broader comparative studies with additional cave-adapted polyploid species and chromosome-level assemblies are needed to separate lineage-specific adaptations from general patterns.
Supplementary Information
Additional file 1.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Zhao Y, Zhang C Endemic fishes of Sinocyclocheilus (Cypriniformes: Cyprinidae) in China—Species diversity, cave adaptation, systematics and zoogeography. Science Press, Beijing. 2009.
- 2Jeffery WR Astyanax mexicanus: a vertebrate model for evolution, adaptation, and development in caves. In Encyclopedia of Caves. 2019. pp 85–93.
- 3Wei L, Ji L, Han S, Xu M, Yang X Construction and validation of a prognostic model of metabolism-related genes driven by somatic mutation in bladder cancer. Front Biosci. 2023;28(10):242. (Landmark edition)10.31083/j.fbl 281024237919060 · doi ↗ · pubmed ↗
- 4Mao T, Liu Y, Svardal H, Vasconcellos M, Yang J, Yang L, He S, Meegaskumbura M Chromosomal restructuring and subgenome divergence drive post-polyploid adaptive diversification in Sinocyclocheilus cavefish. bio Rxiv 2025:2025.2009. 2022.677718.
- 5Kowalko JE, Ma L, Jeffery WR Genome editing in Astyanax mexicanus using transcription activator-like effector nucleases (TALE Ns). J Vis Exp. 2016;112:e 54113.10.3791/54113 PMC 499324027404092 · doi ↗ · pubmed ↗
- 6Ma L, Zhao Y Cavefish of China. In Encyclopedia of caves. Elsevier; 2012. pp. 107–125.