Dramatic response to entrectinib in a rare glioneuronal tumor harboring an NTRK2 fusion
Firas Akrout, Henri Bogumil, Mohamed Dehmani Yedeas, Philipp Sievers, Mehdi Touat, Sameh Achoura

TL;DR
A rare brain tumor in a young woman responded completely to entrectinib after DNA methylation profiling identified an NTRK2 fusion.
Contribution
Demonstrates the efficacy of entrectinib in treating NTRK2 fusion-positive glioneuronal tumors through DNA methylation profiling.
Findings
DNA methylation profiling reclassified the tumor as GTAKA with a KANK1::NTRK2 fusion.
Entrectinib therapy led to a complete radiological response and clinical improvement after 14 months.
The case supports entrectinib as a potential treatment for NTRK fusion–positive glioneuronal tumors.
Abstract
Glioneuronal tumors are rare CNS neoplasms that can exhibit overlapping histological features with embryonal tumors, posing diagnostic and therapeutic challenges. We report a case of a 19-year-old Tunisian woman with a large right frontal tumor and contralateral extension. Initial partial resection suggested CNS neuroblastoma, and the patient underwent chemoradiotherapy with temporary disease control. Upon progression, a second partial resection was followed by DNA methylation profiling, which reclassified the tumor as a glioneuronal tumor with ATRX alteration, kinase fusion, and anaplastic features (GTAKA), harboring a KANK1::NTRK2 fusion. Entrectinib therapy was initiated, leading to a complete radiological response at 14 months, with marked clinical improvement and no serious adverse effects. This case highlights the essential role of DNA methylation profiling in resolving diagnostic…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
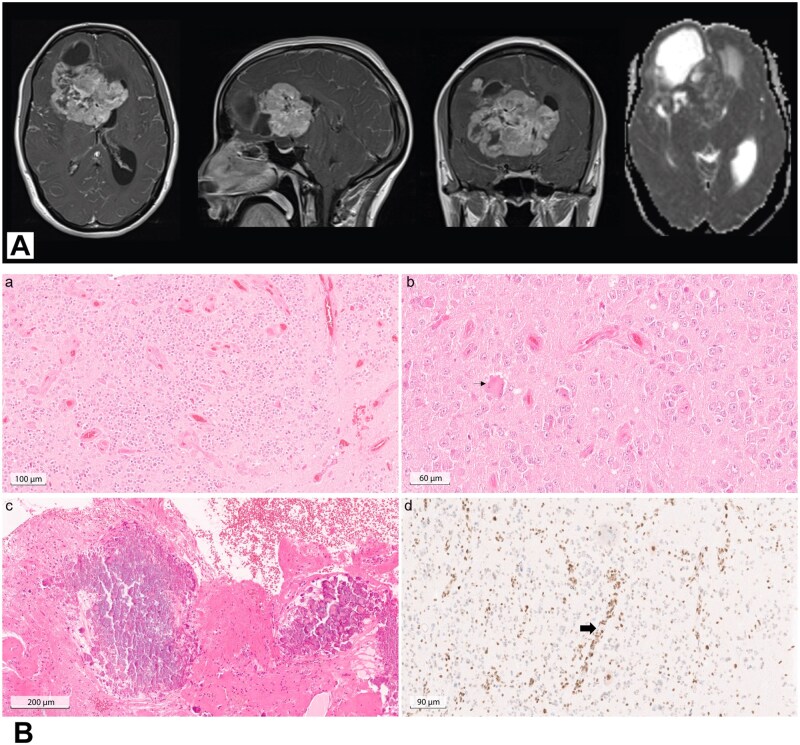 Figure 1
Figure 1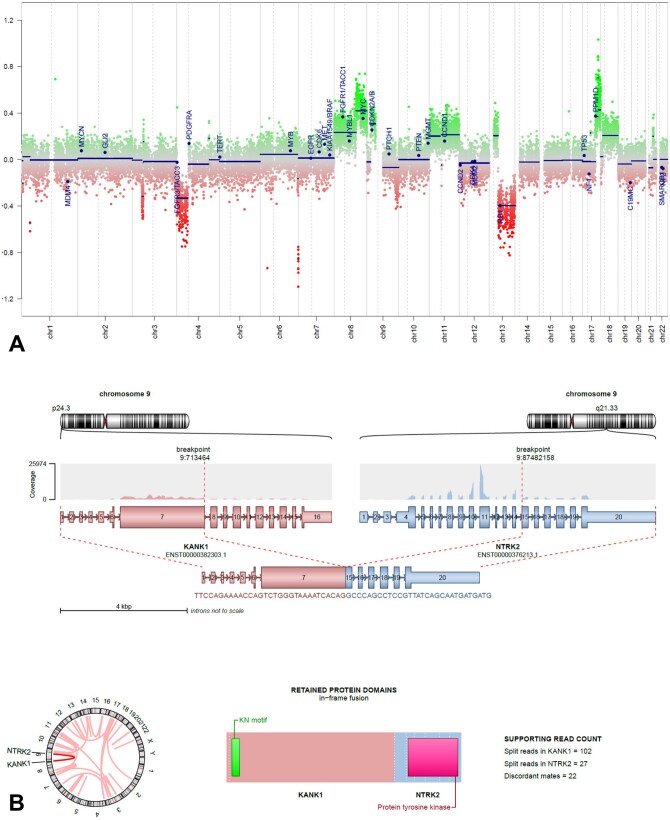 Figure 2
Figure 2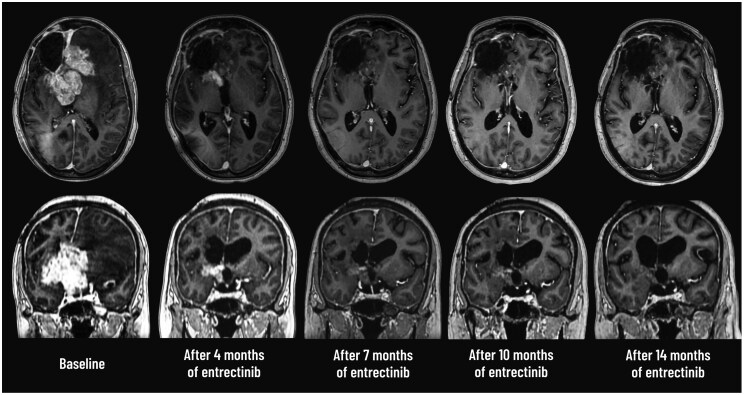 Figure 3
Figure 3| Gene | Consequence | Transcript of variant | Allele frequency | Allele depth | Depth of sequencing | Classification according to ClinGen-CGC-VICC Guidelines | Reference genome |
|---|---|---|---|---|---|---|---|
|
| missense variant | 50.41 | 1169 | 2319 | VUS | hg38 | |
|
| missense variant | 20.8 | 57 | 274 | VUS | hg38 | |
|
| missense variant | 19.67 | 155 | 788 | VUS | hg38 | |
|
| missense variant | 50.35 | 284 | 564 | VUS | hg38 | |
|
| missense variant | 32.91 | 968 | 2941 | VUS | hg38 | |
|
| missense variant | 45.05 | 387 | 859 | VUS | hg38 | |
|
| missense variant | 75.93 | 975 | 1284 | VUS | hg38 | |
|
| missense variant | 46.65 | 746 | 1599 | VUS | hg38 | |
|
| missense variant | 46.31 | 257 | 555 | VUS | hg38 | |
|
| missense variant | n.151275131G>T | chr7: g.151275131G>T | 48.5 | 663 | 1367 | VUS | hg38 |
|
| missense variant | 43.9 | 587 | 1337 | VUS | hg38 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGlioma Diagnosis and Treatment · Neuroblastoma Research and Treatments · Neurofibromatosis and Schwannoma Cases
Introduction
Glioneuronal tumors represent a heterogeneous group of central nervous system (CNS) neoplasms, with emerging molecular classifications reshaping diagnostic paradigms and therapeutic approaches. Among these, Glioneuronal Tumor with ATRX Alteration, Kinase Fusion, and Anaplastic Features (GTAKA) has recently been delineated as a distinct molecular entity. It is defined by a characteristic DNA methylation signature and recurrent gene fusions, predominantly involving the NTRK gene family.1 These oncogenic fusions are key drivers of tumor growth and survival, making TRK proteins attractive targets for precision therapy. Entrectinib, a CNS-penetrant tyrosine kinase inhibitor with potent activity against TRK, ROS1, and ALK fusion proteins, has demonstrated efficacy in NTRK-rearranged solid tumors such as non-small cell lung cancer and secretory carcinomas.2 However, its application in primary CNS tumors, particularly rare glioneuronal subtypes like GTAKA, remains understudied. We present a case of NTRK fusion-driven GTAKA with sustained and rapid clinical and radiological response to entrectinib, illustrating how molecular profiling can identify actionable targets and expand therapeutic options for aggressive glioneuronal tumors.
Patient story
The patient, a 19-year-old Tunisian woman with no past medical history, presented with symptoms of raised intracranial pressure. Neurological examination was unremarkable except for a frontal syndrome characterized by disinhibited behavior. MRI showed a large supratentorial lesion mainly located in the right frontal lobe with contralateral involvement. The enhancing component of the lesion exhibited diffusion restriction. MR spectroscopy demonstrated a clear increase in choline with a decrease of the N-acetyl aspartate (NAA) peak (Figure 1(A)). A partial resection was performed. The initial histological diagnosis, based on morphological and immunohistochemical studies performed in two different expert neuropathology centers, was a primary CNS neuroblastoma (WHO 2016 grade IV).3 The tumor proliferation showed high cellularity and consisted of neurocytic-like cells, which were mostly positive for Olig2 and synaptophysin and partially positive for Lin28 and Neu-N. MRI at 3 months postoperatively showed a neoplastic residue in the tumor bed and disease progression. Based on the original diagnosis, three cycles of EP (cisplatin (100 mg/m^2^/d) and etoposide (120 mg/m^2^/d)) were administered. Chemotherapy was interrupted due to grade 3 neutropenia after the second and third courses. Concomitantly, the patient received whole brain irradiation (30.6 Gy) with a boost to the initial tumor bed (23.4 Gy). Follow-up MRI scans showed a significant partial response, with an estimated 59.2% reduction in tumor size. After a period of disease stability for 26 months from therapy discontinuation, the patient developed symptoms of elevated intracranial pressure and cognitive impairment. Imaging revealed significant progression of the residual tumor and associated hydrocephalus, prompting a repeat partial resection, placement of a ventriculoperitoneal shunt, and a subsequent re-evaluation of the primary tumor’s pathology.
(A) Preoperative cranial MRI of the patient (from left to right: axial, coronal and sagittal T1-weighted with gadolinium injection, axial apparent diffusion coefficient). There is a solid intra-axial mass within the right frontal lobe with contralateral involvement. The enhancing component of the lesion shows diffusion restriction. (B) HE slides of the primary tumor showed a cell dense glioneuronal tumor with microvascular proliferation (a, scale bar 100 µm). Multinuclear cells (b, small arrow, scale bar 60 µm) as well as calcification (c, scale bar 200 µm) could be observed. ATRX immunohistochemistry (d, scale bar 90 µm) revealed near complete loss of nuclear staining in tumor cells in comparison to non-neoplastic cells such as blood vessels (large arrow).
Molecular tumor board
Tumor samples were referred for an expert opinion to Heidelberg. Histological analyses showed a cell-dense glioneuronal tumor with elevated mitotic activity and microvascular proliferation (Figure 1(B)). Adjacent CNS tissue revealed hemosiderin pigment, fresh hemorrhages and focal microinfarction.
For further classification, DNA methylation analysis was performed using Illumina XX methylome array, with analysis using the diagnostic Heidelberg pipeline.4 The methylation data showed a match (0,90) with the methylation class super-family Adult Type Diffuse Gliomas with the highest score (0,89) for the methylation class “Glioneuronal tumor with ATRX alteration, kinase fusion and anaplastic features (novel)”. Copy number variation profile revealed several chromosomal gains (chromosomes 8, 11 and 17) and losses (chromosomes 4 and 13) as well as a balanced CDKN2A/B status (Figure 2(A)). MGMT promoter was predicted unmethylated. Consistent with the results of DNA methylation analysis, ATRX immunohistochemistry was performed (mouse monoclonal, clone BSB-108, dilution 1:2000, Bio SB, Santa Barbara, CA, USA) and showed a near complete loss of ATRX nuclear expression in tumor cell nuclei compared to non-neoplastic cells (Figure 1(B)).
(A) Copy number variation profile generated from DNA methylation data shows several chromosomal gains and losses. (B) Visualization of the detected KANK1::NTRK2 fusion including chromosomal breakpoints and involved exons of both fusion partners.
As tumors of the methylation class “Glioneuronal tumor with ATRX alteration, kinase fusion and anaplastic features (novel)” usually harbor kinase fusions,1 RNA sequencing was performed using a previously described diagnostic pipeline.5 An oncogenic in-frame KANK1::NTRK2 fusion was detected with preserved protein tyrosine kinase domain of NTRK2 (Figure 2(B)). In conclusion of histology and molecular analyses, an integrated diagnosis of “Glioneuronal tumor with ATRX alteration, kinase fusion and anaplastic features (GTAKA)” was established.
To further investigate the ATRX staining, a DNA panel sequencing was performed using a previously described diagnostic workflow.6 Analyses showed two missense variants in ATRX, which were not yet described in the literature (Table 1), fitting to the diagnosis.
Patient update
Given the identification of an oncogenic NTRK2 fusion, the patient was initiated on the selective TRK inhibitor entrectinib at a daily dose of 600 mg. Within the first three months of therapy, she achieved full restoration of cognitive function and maintained a Karnofsky performance status of 90%, allowing her to resume her university studies and reclaim independent daily activities without assistance. Successive follow-up MRIs at 4, 7, and 10 months showed a progressive reduction in tumor volume, resulting in complete radiological regression at 14 months and confirming a durable response to entrectinib (Figure 3). The treatment was well tolerated, demonstrating a favorable safety profile. Aside from a brief episode of dysgeusia and weight gain, no other toxicities were observed, and laboratory parameters remained unremarkable. At the time of this report, the overall treatment duration and follow-up are 14 months and 8 days.
Radiological evolution of the tumor under entrectinib therapy. Axial, and coronal post-contrast T1-weighted MRI sequences obtained at baseline, and after 4, 7, 10 and 14 months of entrectinib treatment (from left to right). A marked and progressive reduction in the volume of the enhancing tumor is observed over time, indicating a sustained radiological response.
Discussion
Accurate classification of CNS neoplasms remains challenging in neuropathology, particularly when tumors exhibit overlapping histological and immunophenotypic features. As illustrated by our case, glioneuronal tumors such as GTAKA can histologically be misclassified as CNS neuroblastoma, or vice versa, owing to shared morphological characteristics (small round blue cell appearance) and neuronal marker expression (synaptophysin, NeuN).8–10 This diagnostic ambiguity carries profound clinical consequences: embryonal tumors like neuroblastoma conventionally require aggressive multimodal chemotherapy, while NTRK fusion–driven glioneuronal tumors may derive durable benefit from targeted inhibitors such as entrectinib. Misclassification can therefore expose patients to unnecessary toxicities from cytotoxic regimens while delaying access to effective biology‑driven therapies.
This dilemma underscores the imperative of DNA methylation profiling in CNS tumors with ambiguous or heterogeneous histology, particularly those displaying mixed glial/neural or glial/embryonal components. By comparing a tumor’s genome-wide methylation pattern to established reference profiles, this method accurately identifies the tumor type, determines its WHO grade, and reveals targetable molecular alterations, as shown in our case. Our findings align with a growing consensus that methylation analysis should be integrated as a first‑line diagnostic modality for histologically equivocal CNS neoplasms, thereby enabling tailored, precision‑based management.11
Entrectinib is a potent, orally bioavailable, CNS-penetrant small-molecule inhibitor designed to selectively target tyrosine receptor kinases involved in oncogenic gene fusions. Specifically, it inhibits the tropomyosin receptor kinases TRKA, TRKB, and TRKC (encoded by NTRK1, NTRK2, and NTRK3, respectively), as well as ROS1 and ALK fusion proteins. These oncogenic fusions drive tumorigenesis by promoting cell proliferation and survival. In August 2019, entrectinib received FDA approval for adult and pediatric patients ≥12 years with NTRK fusion-positive solid tumors and for ROS1-positive non-small cell lung cancer, representing a milestone in the adoption of molecularly guided cancer therapies. Other TRK inhibitors include the first-in-class agent larotrectinib, approved in 2018, and several next-generation inhibitors currently in development, such as selitrectinib (LOXO-195), repotrectinib (TPX-0005), and taletrectinib (DS-6051b), which aim to overcome resistance to first-generation agents.
Entrectinib has demonstrated robust efficacy across a range of NTRK fusion–positive solid tumors, including non-small cell lung cancer, secretory breast carcinoma, and salivary gland tumors.2 Its ability to penetrate the blood–brain barrier makes it particularly relevant for the treatment of primary CNS tumors, which often present therapeutic challenges due to limited drug delivery to the CNS.12 In the phase 1/2 STARTRK‑NG trial, entrectinib demonstrated rapid and durable activity against pediatric intracranial and extracranial solid tumors harboring NTRK1/2/3 or ROS1 fusions.13
While clinical benefit from NTRK inhibitors in gliomas and glioneuronal tumors with NTRK fusions is documented in several case reports and small series, prospective data remain limited, and the target holds an ESCAT IIB score, pending validation of efficacy in larger clinical trials or registries.14^,^15 The literature specifically addressing NTRK fusion–positive glioneuronal tumors treated with TRK inhibitors is particularly sparse, with only four cases published to date. Two cases, harboring ARHGEF2–NTRK1 and BCAN–NTRK1 fusions, were treated with entrectinib and achieved partial responses, with follow-up durations of 9 and 6 months, respectively.16^,^17 The other two cases, involving STRN1–NTRK2 and MEF2D–NTRK1 fusions, were treated with larotrectinib and demonstrated complete responses maintained at 11 and 13 months of follow-up, respectively.18^,^19 Notably, all previously reported cases involved minimal residual tumor volume at the time of TRK inhibitor initiation, whereas our patient received entrectinib in the context of a substantial 6 cm residual tumor, highlighting the potential of TRK inhibition even in cases with high tumor volume. Our findings further support the importance of testing for NTRK fusions and the potential therapeutic value of targeted treatment in this context.
To our knowledge, this is the first reported case of a GTAKA showing a rapid and profound clinical and radiological response to entrectinib. The near-complete regression achieved within 16 weeks underscores the rapid therapeutic efficacy of TRK inhibition, even in tumors with high initial volume. This case reinforces the critical role of early molecular profiling in identifying actionable alterations such as NTRK fusions and highlights the promise of precision medicine in the management of rare and aggressive CNS tumors.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bogumil H , Sill M, Schrimpf D, et al. Glioneuronal tumor with ATRX alteration, kinase fusion and anaplastic features (GTAKA): a molecularly distinct brain tumor type with recurrent NTRK gene fusions. Acta Neuropathol. 2023;145:667–680.36933012 10.1007/s 00401-023-02558-0PMC 10119244 · doi ↗ · pubmed ↗
- 2Doebele RC , Drilon A, Paz-Ares L, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020;21:271–282.31838007 10.1016/S 1470-2045(19)30691-6PMC 7461630 · doi ↗ · pubmed ↗
- 3Louis DN , Perry A, Reifenberger G, et al. The 2016 world health organization classification of tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131:803–820.27157931 10.1007/s 00401-016-1545-1 · doi ↗ · pubmed ↗
- 4Capper D , Jones DTW, Sill M, et al. DNA methylation-based classification of Central nervous system tumours. Nature. 2018;555:469–474.29539639 10.1038/nature 26000 PMC 6093218 · doi ↗ · pubmed ↗
- 5Stichel D , Schrimpf D, Casalini B, et al. Routine RNA sequencing of formalin-fixed paraffin-embedded specimens in neuropathology diagnostics identifies diagnostically and therapeutically relevant gene fusions. Acta Neuropathol. 2019;138:827–835.31278449 10.1007/s 00401-019-02039-3 · doi ↗ · pubmed ↗
- 6Sahm F , Schrimpf D, Jones DTW, et al. Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol. 2016;131:903–910.26671409 10.1007/s 00401-015-1519-8 · doi ↗ · pubmed ↗
- 7Horak P , Griffith M, Danos AM, et al. Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): joint recommendations of clinical genome resource (Clin Gen), cancer genomics consortium (CGC), and variant interpretation for cancer consortium (VICC). Genet Med. 2022;24:986–998.35101336 10.1016/j.gim.2022.01.001PMC 9081216 · doi ↗ · pubmed ↗
- 8Tauziède-Espariat A , Figarella-Branger D, Métais A, et al. CNS neuroblastoma, FOXR 2-activated and its mimics: a relevant panel approach for work-up and accurate diagnosis of this rare neoplasm. Acta Neuropathol Commun. 2023;11:43.36918978 10.1186/s 40478-023-01536-7PMC 10012567 · doi ↗ · pubmed ↗