Characterization of Verbesina encelioides (Asteroideae, Asteraceae) Chloroplast Genome and Phylogenetic Insights
Rushan Yan, Abdullah, Jingjing Jia, Madiha Islam, Hui Li, Mengyang Liu, Bartholomew Yir‐erong, Xiaoxuan Tian

TL;DR
This study reports the first complete chloroplast genome of Verbesina encelioides, providing insights into its structure, gene content, and evolutionary relationships within the Asteraceae family.
Contribution
The first de novo assembled and annotated chloroplast genome of Verbesina encelioides is presented, offering new phylogenetic and genetic insights.
Findings
The chloroplast genome of Verbesina encelioides is 152,213 bp with a typical quadripartite structure and 112 unique genes.
Comparative analysis with Verbesina alternifolia shows high structural and sequence conservation.
Codon usage is biased toward A/T-ending codons, and 19 chloroplast genes show codons under positive selection.
Abstract
Verbesina encelioides (Cav.) Benth. & Hook.f. ex A.Gray (Asteroideae, Asteraceae) is a widespread annual herb native to southwestern North America that has naturalized globally. Here, we report the first de novo assembly and comprehensive annotation of the complete chloroplast (cp) genome of V. encelioides , generated using Illumina NovaSeq sequencing. The circular genome is 152,213 bp and exhibits the characteristic quadripartite structure, comprising a large single‐copy (83,911 bp) region, a small single‐copy (18,248 bp) region, and two inverted repeat regions (25,027 bp each). The genome encodes 112 unique genes, including 79 protein‐coding genes, 29 tRNAs, and four rRNAs, with 16 genes duplicated in the IRs. Comparative analysis with Verbesina alternifolia revealed high structural conservation regarding gene content and arrangement, codon usage, amino acid frequency, and simple…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 FIGURE 1
FIGURE 1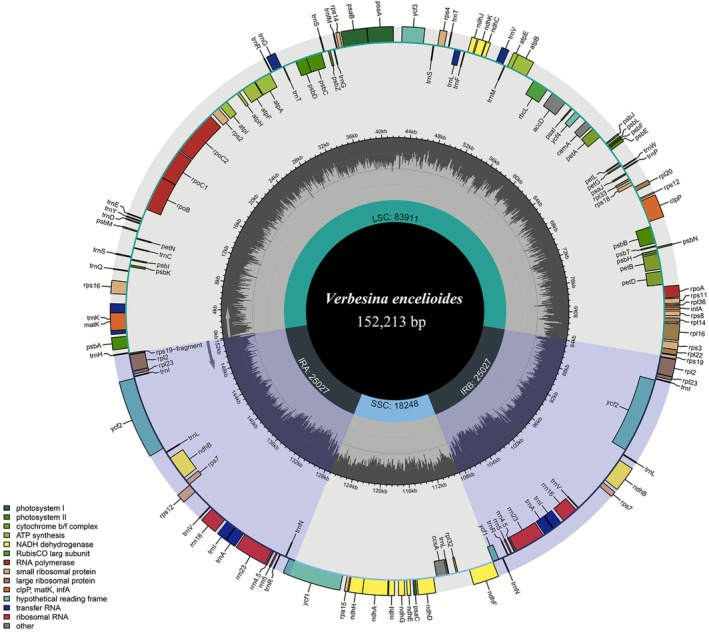 FIGURE 2
FIGURE 2 FIGURE 3
FIGURE 3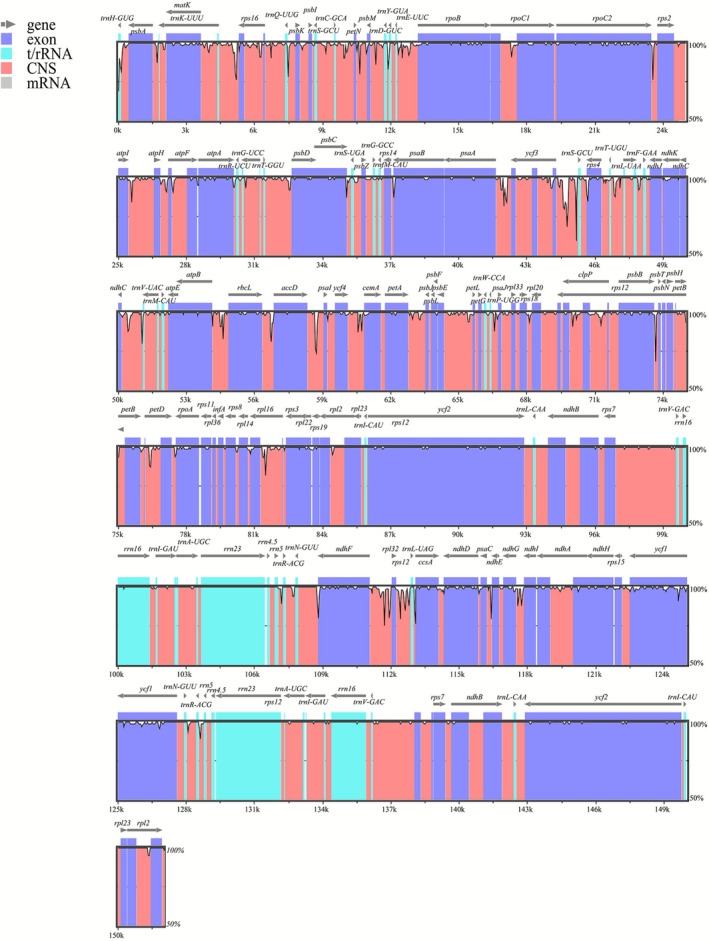 FIGURE 4
FIGURE 4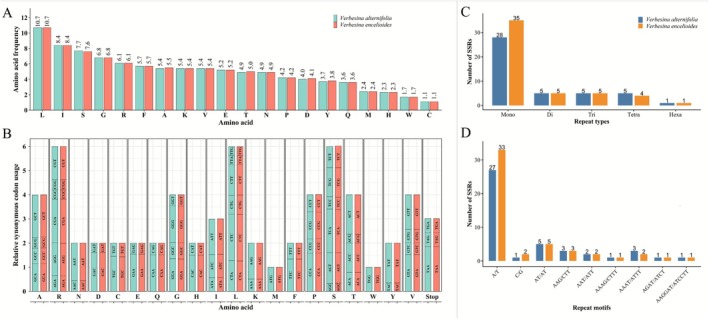 FIGURE 5
FIGURE 5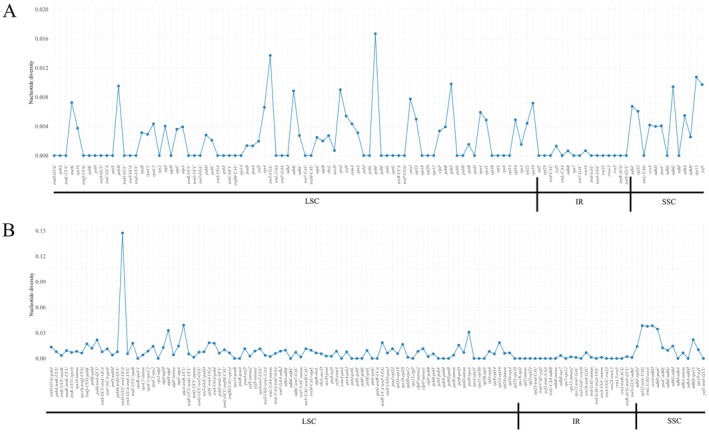 FIGURE 6
FIGURE 6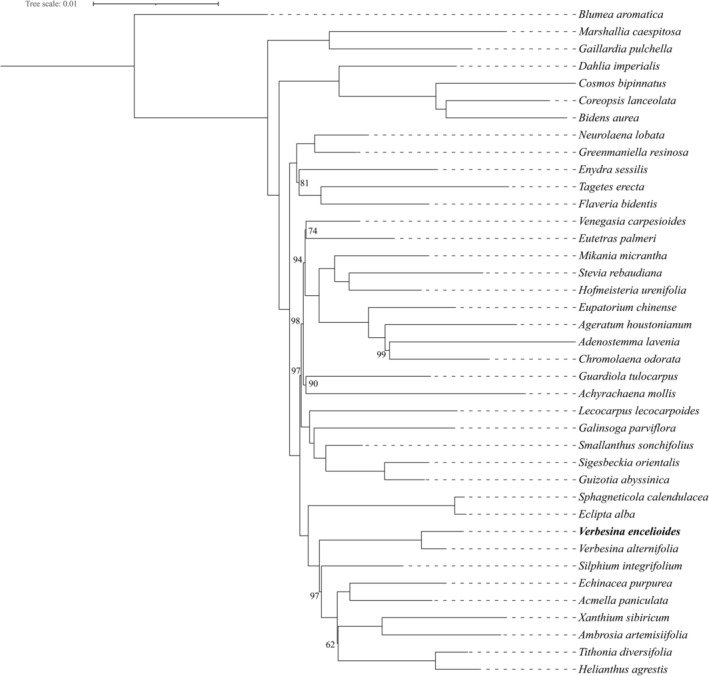 FIGURE 7
FIGURE 7| Characteristics |
|
|
|---|---|---|
| Total size (bp) | 152,050 | 152,213 |
| LSC length (bp) | 83,725 | 83,911 |
| SSC length (bp) | 18,245 | 18,248 |
| IR length (bp) | 25,040 | 25,027 |
| Unique genes | 112 | 112 |
| Protein‐coding genes | 79 | 79 |
| tRNA genes | 29 | 29 |
| rRNA genes | 4 | 4 |
| Duplicate genes | 16 | 16 |
|
| ||
| Total | 37.7 | 37.7 |
| LSC | 35.8 | 35.8 |
| SSC | 31.4 | 31.5 |
| IR | 43.1 | 43.1 |
| CDS | 38 | 37.9 |
| rRNA | 55.2 | 55.2 |
| tRNA | 53 | 53.1 |
| All gene | 39.5 | 39.5 |
| Category for genes | Group of genes | Name of genes | Amount |
|---|---|---|---|
| Self‐replication | Large subunit of ribosome |
| 11 |
| Small subunit of ribosome |
| 13 | |
| DNA dependent RNA polymerase |
| 4 | |
| rRNA genes |
| 8 | |
| tRNA genes |
| 36 | |
| Photosynthesis | Photosystem I |
| 5 |
| Photosystem II |
| 15 | |
| NADPH dehydrogenase |
| 12 | |
| Cytochrome b/f complex |
| 6 | |
| Subunits of ATP synthase |
| 6 | |
| Photosystem I assembly proteins |
| 2 | |
| Large subunit of Rubisco |
| 1 | |
| Other genes | Protease |
| 1 |
| Maturase |
| 1 | |
| Envelop membrane protein |
| 1 | |
| Subunit of Acetyl‐CoA‐carboxylase |
| 1 | |
| C‐type cytochrome synthesis gene |
| 1 | |
| Translation initiation factor |
| 1 | |
| Conserved open reading frames |
| 3 |
| Substitution type | LSC region | SSC region | IR region |
|---|---|---|---|
| A/G | 115 | 45 | 7 |
| G/T | 73 | 24 | 3 |
| A/C | 63 | 20 | 3 |
| C/T | 109 | 43 | 5 |
| C/G | 27 | 5 | 2 |
| A/T | 42 | 32 | 0 |
| Ts | 224 | 88 | 12 |
| Tv | 205 | 81 | 8 |
| Ts/Tv | 1.09 | 1.08 | 1.5 |
| Serial number | Region | Nucleotide diversity | Number of substitutions | Number of indels | Length (excluding indels) | Alignment length | Missing data (%) |
|---|---|---|---|---|---|---|---|
|
| |||||||
| 1 |
| 0.01667 | 2 | 0 | 120 | 120 | 0 |
| 2 |
| 0.01075 | 3 | 0 | 279 | 279 | 0 |
| 3 |
| 0.0098 | 1 | 0 | 102 | 102 | 0 |
| 4 |
| 0.00972 | 49 | 6 | 5043 | 5049 | 0.12 |
| 5 |
| 0.00952 | 1 | 0 | 105 | 105 | 0 |
|
| |||||||
| 6 |
| 0.14729 | 19 | 1 | 129 | 130 | 0.77 |
| 7 |
| 0.03906 | 5 | 4 | 128 | 132 | 3.0 |
| 8 |
| 0.03846 | 25 | 36 | 650 | 686 | 5.2 |
| 9 |
| 0.03831 | 10 | 14 | 261 | 275 | 5.1 |
| 10 |
| 0.03774 | 4 | 23 | 106 | 129 | 17.8 |
- —National Natural Science Foundation of China10.13039/501100001809
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Sesquiterpenes and Asteraceae Studies · Plant Diversity and Evolution
Introduction
1
Asteraceae, one of the largest and most ecologically successful angiosperm families, comprises approximately 1620 genera and 34,000 species distributed across 16–17 subfamilies (Zhang, Yang, et al. 2024; WFO 2025). Members of Asteraceae occur worldwide, including Antarctica, and occupy diverse habitats. They display remarkable morphological and ecological plasticity, comprising annual and perennial herbs, shrubs, trees, vines, and succulents (Funk 2009; Susanna and Baldwin 2020; Roeble et al. 2024).
The genus Verbesina L., within the subtribe Verbesininae of the tribe Heliantheae, subfamily Asteroideae, is endemic to the Americas and comprises 351 species (POWO 2025). To date, only one species in the genus, Verbesina alternifolia (L.) Britton ex Kearney, has a published chloroplast (cp) genome (Tomasello et al. 2024). Verbesina encelioides (Cav.) Benth. & Hook.f. ex A.Gray, native to southwestern North America, has naturalized in disturbed areas globally. This annual herb thrives in temperate to subtropical biomes and is well adapted to arid and sandy soils (Gorja and Bandla 2024). Owing to its aggressive spread, strong ecological competitiveness, and invasive behavior in many regions, V. encelioides has become a focal species in studies of biological invasion, abiotic‐stress adaptation, and ecological risk assessment (Menghani 2008; Singh 2010; Mehal et al. 2023). However, the cp genome of this species remains unavailable, limiting our understanding of its dispersal mechanisms and evolutionary history. Characterizing the complete cp genome would enable phylogeographic analyses, facilitate the development of robust molecular markers, and support invasive species management strategies. Moreover, V. encelioides exhibits important pharmacological activities and is widely used in traditional medicine (Kataria et al. 2025), making genomic resources valuable for both ecological and medicinal research applications.
The cp genome typically exhibits a conserved quadripartite structure, comprising a large single‐copy (LSC) region, a small single‐copy (SSC) region, and two inverted repeats (IRa and IRb) (Palmer 1985; Daniell et al. 2016). Owing to its moderate polymorphism and predominantly maternal inheritance in angiosperms, the cp genome serves as a valuable resource for phylogenetic reconstruction, population genetics, DNA barcoding, and conservation studies (Daniell et al. 2016; Zhang, Huang, et al. 2024; Zeng et al. 2025). Several mutations have been reported in cp genomes, including substitutions, insertions–deletions, inversions, and contraction or expansion of inverted repeats, which can alter gene content (Daniell et al. 2016; Zhang, Huang, et al. 2024; Abdullah, Haram, et al. 2025; Yan et al. 2025; Abdullah, Li, et al. 2025).
To expand genomic resources for Verbesina, we sequenced and de novo assembled the V. encelioides cp genome to characterize its genetic features, conduct comparative analyses with V. alternifolia , identify polymorphic loci with potential phylogenetic utility, assess signatures of molecular evolution, and reconstruct its phylogenetic relationships within the tribe. This study provides the first complete cp genomic resource for V. encelioides , filling a critical data gap and offering a molecular foundation for future evolutionary, ecological, and invasion biology research on this globally expanding species.
Materials and Methods
2
Sample Collection, DNA Extraction, and Sequencing
2.1
V. encelioides was collected from District Nowshera, Khyber Pakhtunkhwa Province, Pakistan (34°04′38.68″ N, 71°90′97.17″ E) and identified by Dr. Abdul Majid, Assistant Professor at Hazara University, Mansehra. The voucher specimen was submitted to the herbarium of Hazara University, Mansehra, under accession number HUP‐17585. Permission was not required from the national or local authorities for plant collection and utilization in research. A photograph of the plant is provided in Figure 1. Leaves were dried using silica gel for DNA extraction. DNA was extracted from 30 mg of dried leaf tissue using the Plant Genomic DNA Kit (TIANGEN BIOTECH, Beijing, China) with the following modifications: (a) tissue was homogenized in 1.5 mL of GP1 buffer, and the resulting lysate was divided into two 1.5‐mL microcentrifuge tubes for parallel processing; (b) the incubation at 65°C was extended to 60 min; (c) supernatants from both tubes were combined and loaded onto a single DNA‐binding column (CB3); and (d) final elution was performed with 95 μL of elution buffer after a 40‐min incubation at room temperature.
Verbesina encelioides in its natural habitat. The photograph, taken by Dr. Abdullah, highlights several inflorescences of V. encelioides . Given the dense surrounding vegetation and the coexistence of other plant species, the image emphasizes clearly distinguishable flowers to facilitate accurate identification.
DNA quality and concentration were evaluated using an Agilent 5400 System (Agilent Technologies), which confirmed high‐molecular‐weight DNA at 19.74 ng/μL (total yield ≈ 1.8 μg). A library with a 350‐bp insert size was prepared, and paired‐end sequencing (2 × 150 bp) was performed on an Illumina NovaSeq 6000 platform at Novogene (Tianjin, China).
Chloroplast Genome Assembly and Annotation
2.2
We assessed raw read quality using Fastp v1.0.1 (Chen 2025). Reads containing > 10% ambiguous nucleotides (N) or > 50% bases with quality score (Q) ≤ 5 were filtered out. The resulting clean reads, which had Q20 and Q30 scores of 99% and 96%, respectively, were used for de novo cp genome assembly using GetOrganelle v1.7.5^+^ (Jin et al. 2020) with k‐mer sizes of 21, 45, 65, 85, and 105.
Genome annotation was performed using GeSeq v2.03 (Tillich et al. 2017), while transfer RNA genes were further verified using ARAGORN v1.2.38 (Laslett and Canback 2004) and tRNAscan‐SE v2.0.7 (Chan and Lowe 2019). Start and stop codons of protein‐coding sequences (CDS) were manually verified and corrected in Geneious Prime 2025, based on comparisons with Verbesina alternifolia (PP_639077), Eclipta alba (NC_039774), and Silphium integrifolium (NC_068130).
Comparative Genomics, Amino Acid Frequency, and Codon Usage Analysis
2.3
The cp genomes of V. alternifolia and V. encelioides were compared using Geneious Prime 2025 and the mVISTA tool (Frazer et al. 2004) in Shuffle‐LAGAN mode. Amino acid frequency and relative synonymous codon usage (RSCU) were analyzed using custom Python scripts (Abdullah, Fatima, et al. 2025).
Analysis of Nucleotide Substitutions and Microsatellites
2.4
To analyze nucleotide substitutions, the LSC, SSC, and IR regions of V. alternifolia and V. encelioides were extracted and aligned pairwise. Transition and transversion substitutions were identified, and their ratios were calculated using a custom Python script (Script 1). Simple sequence repeats (SSRs) were identified using MISA‐web (https://webblast.ipk‐gatersleben.de/misa/) (Beier et al. 2017). Search parameters were set to a minimum of 10 repeat units for mononucleotides, 5 for dinucleotides, 4 for trinucleotides, and 3 for tetranucleotides, pentanucleotides, and hexanucleotides.
Analysis of Adaptive Evolution
2.5
To evaluate signatures of episodic diversifying selection, HyPhy v2.5.7 (Pond et al. 2005) was used to implement the Mixed Effects Model of Evolution (MEME) (Murrell et al. 2012). Codon‐based alignments were generated separately for each protein‐coding gene using MUSCLE v5 (Edgar 2022), and terminal stop codons were removed in Geneious prior to analysis. A total of 15 complete chloroplast genomes representing 15 closely related Asteraceae species were included (Table S1). For each gene, a maximum‐likelihood phylogeny was reconstructed using FastTree v2 (Price et al. 2009) with default settings, as required for MEME to infer branch‐specific variation in selective pressures.
Analysis of Nucleotide Polymorphism and Phylogenetic
2.6
Nucleotide diversity (Pi) for all cp genome regions, including introns, CDS, and intergenic spacers (IGS) regions, was calculated using CPStools (L. Huang et al. 2024).
We retrieved 38 species from the National Centre for Biotechnology Information (NCBI) belonging to distinct genera of tribe Heliantheae and aligned them with Verbesina species, including Blumea aromatica (NC_069835) from tribe Inuleae as the outgroup, using MAFFT (Nakamura et al. 2018). The phylogenetic tree was reconstructed from whole‐genome alignments using the maximum likelihood (ML) method implemented in IQ‐TREE v3.0.1 (Wong et al. 2025) with automated model selection (ModelFinder), SH‐aLRT branch support, and 1000 ultrafast bootstrap replicates with the ‐‐bnni option, following a previous description (Abdullah, Haram, et al. 2025). The resulting trees were inferred under the BIC‐selected model (GTR + F) and visualized using the Interactive Tree of Life platform (iTOL) (Letunic and Bork 2024; http://itol.embl.de/).
Results and Discussion
3
Characterization of the Chloroplast Genome of
Verbesina encelioides
3.1
Sequencing V. encelioides generated approximately 5.44 GB of data, yielding 7.8 million paired‐end clean reads (150 bp each). The cp genome was assembled de novo with an exceptionally high average coverage depth of 2281× from 2.3 million cp‐specific reads. The complete cp genome measured 152,213 bp and exhibited the typical quadripartite structure (Figure 2), comprising the LSC region (83,911 bp), the SSC region (18,248 bp), and two IR regions (IRa/IRb; 25,027 bp each). Such high coverage ensures reliable assembly and supports robust downstream comparative analyses. The quadripartite organization observed here is characteristic of most land plant cp genomes and reflects the evolutionary conservation of this structural arrangement across diverse angiosperm lineages (Abdullah, Haram, et al. 2025; Y. Huang et al. 2025; Xing et al. 2025).
Circular map depicting the chloroplast genome of V. encelioides . Genes transcribed clockwise are shown inside the circle, whilst those transcribed anticlockwise are shown outside the circle. Genes are color‐coded based on function. Darker gray in the inner circle represents GC content throughout the genome.
The overall GC content was 37.7%, with marked regional variation: LSC (35.8%), IRs (43.1%), and SSC (31.5%). Among gene classes, GC content was highest in rRNA genes (55.2%), followed by tRNAs (53.1%) and protein‐coding sequences (CDS) (37.9%). Such GC variation likely reflects differential selective pressures, with higher GC content in IR regions contributing to structural stability and conservation. The genome showed high similarity in length and GC content to other Verbesina species (Table 1) and to previously reported species from the tribe Heliantheae and the subfamily Asteroideae (Abdullah et al. 2021; Mahai et al. 2024; Karimov et al. 2025; Xue et al. 2025). This GC profile aligns with patterns observed across Asteroideae in genera such as Artemisia and Blumea (Iram et al. 2019; Abdullah et al. 2021), suggesting tribe‐level evolutionary constraints on GC content that are conserved across diverse ecological contexts rather than being species‐specific adaptations.
The cp genome contained 112 unique genes: 79 CDS genes, 29 transfer RNA (tRNA) genes, and four ribosomal RNA (rRNA) genes. Sixteen genes were duplicated within the IR regions, including five CDS genes (ndhB, rpl2, rpl23, rps7, and ycf2), four rRNA genes (rrn16S, rrn23S, rrn4.5S, and rrn5S), and seven tRNA genes (trnA‐UGC, trnI‐GAU, trnL‐CAA, trnN‐GUU, trnR‐ACG, trnV‐GAC, trnI‐CAU) (Table 2). These duplicated genes enhance genome stability by providing redundancy for essential functions.
Eighteen genes contained introns: 12 CDS (atpF, petB, rps16, rps12, rpl2, ndhA, rpl16, ndhB, rpoC1, ycf3, petD, clpP) and six tRNAs (trnV‐UAC, trnK‐UUU, trnG‐UCC, trnI‐GAU, trnL‐UAA, trnA‐UGC). Two CDS genes (ycf3 and clpP) contained two introns each, whilst the remaining 10 had a single intron (Figure 3A). Notably, rps12 was trans‐spliced, with exon 1 in the LSC region and exons 2 and 3 in the IR regions (Figure 3B). This trans‐splicing reflects a conserved mechanism requiring precise coordination during RNA processing. Gene content and intron distribution were consistent with previous reports for tribe Heliantheae and subfamily Asteroideae (Abdullah et al. 2021; Mahai et al. 2024; Karimov et al. 2025; Xue et al. 2025), indicating a highly conserved cp genome structure within this lineage. This conservation is maintained across both invasive and non‐invasive species, demonstrating that invasive properties have not driven structural rearrangements or alterations in gene content within the cp genome. This finding further supports the hypothesis that traits conferring invasiveness are likely governed by nuclear rather than chloroplast genes.
Gene structure map of cis‐ and trans‐spliced genes in the V. encelioides chloroplast genome. (A) Cis‐spliced genes. (B) Trans‐spliced genes.
Comparative Genomics, Amino Acid Frequency, Codon Usage, and Simple Sequence Repeats Analysis
3.2
Comparative genomic analysis between V. encelioides and V. alternifolia revealed high conservation. Intergenic spacers and intron regions were more variable than coding sequences (Figure 4). Most CDS regions were highly conserved, whereas rRNA genes were virtually invariant, reflecting strong purifying selection on coding regions. The invariant nature of rRNA genes underscores their essential role in ribosome assembly and protein synthesis, where mutations are likely deleterious. Similar patterns have been reported in Artemisia, Blumea, and other Asteroideae taxa (Iram et al. 2019; Abdullah et al. 2021; Xing et al. 2025), as well as in dicot families such as Solanaceae, Phyllanthaceae, and Malvaceae (Abdullah, Mehmood, et al. 2020; Abdullah et al. 2021; Rehman et al. 2021). Within Heliantheae, this pattern suggests evolutionary pressures favoring stability in core photosynthetic and translational machinery despite ecological diversification. Notably, the absence of invasiveness‐specific positive selection in the cp genome (discussed in the adaptive evolution section) further supports the hypothesis that traits underlying the invasive success of V. encelioides are likely encoded in the nuclear genome rather than the chloroplast.
Global alignment of chloroplast genomes of V. alternifolia and V. encelioides . We use V. alternifolia as a reference. The x‐axis represents the coordinates in the chloroplast genome. The y‐axis shows the average percent identity of the aligned regions, ranging from 50% to 100%.
Amino acid frequency analysis showed leucine was the most abundant, whereas cysteine was the least encoded (Figure 5A). This reflects the hydrophobic nature of many cp proteins, especially those involved in photosynthesis, and the reducing environment of the cp stroma, which disfavors disulfide bond formation. Codon usage analysis revealed strong bias toward codons ending in A or T at the third position (RSCU > 1), whereas C‐ or G‐ending codons had RSCU < 1 (Figure 5B). This A/T‐ending codon bias is consistent with the high AT content of the cp genome and may influence tRNA abundance and translation efficiency. Similar patterns have been reported in other Asteroideae and plant lineages (Iram et al. 2019; Abdullah, Mehmood, et al. 2020; Abdullah et al. 2021; Rehman et al. 2021).
Analysis of amino acid frequency, codon usage, and simple sequence repeats (SSRs) in the chloroplast genomes of V. alternifolia and V. encelioides . (A) Amino acid frequency distribution, with amino acid types on the x‐axis and their encoded frequency on the y‐axis. (B) Relative synonymous codon usage (RSCU), where the x‐axis indicates amino acids and bar height represents the RSCU value for each species; codons are labeled inside bars. (C) Identified SSR types. (D) SSR motif compositions.
Simple sequence repeats (SSRs) were predominantly mononucleotide repeats (28–35 per genome), followed by di‐ and trinucleotide repeats (Figure 5C), whereas pentanucleotide repeats were absent and hexanucleotide repeats were extremely rare (1 locus) (Figure 5C,D). Most SSRs were composed of A/T‐rich motifs (27–33), fully reflecting the genome‐wide AT bias. These patterns are consistent with previous reports in Blumea and Artemisia (Iram et al. 2019; Abdullah et al. 2021). The prevalence of A/T‐rich SSRs reflects the genome's compositional bias and suggests that slipped‐strand mispairing occurs preferentially in AT‐rich regions. These SSR loci may serve as valuable molecular markers for population genetic studies and for tracing the invasion biogeography of V. encelioides across its expanding global range.
Nucleotide Substitutions
3.3
Pairwise comparison revealed more substitutions and a higher transition‐to‐transversion (Ts/Tv) ratio in the IR regions (1.5) compared to LSC (1.09) and SSC (1.08) (Table 3). The most frequent substitutions were A/G and C/T changes. Although reported Ts/Tv ratios vary in cp genomes, most studies indicate values ≤ 1 (Abdullah et al. 2019), with some reporting ratios > 1 (Cao et al. 2018; Abdullah, Henriquez, et al. 2020). Elevated Ts/Tv ratios in IR regions may reflect sequence context and secondary structure constraints that favor certain mutation types.
Evaluation of Adaptive Evolution
3.4
MEME analysis identified 62 codon sites under episodic diversifying selection distributed across 19 chloroplast genes: atpB, ccsA, clpP, ndhD, ndhI, psaB, psbB, rpl14, rpoB, rpoC1, rps8, ycf3, accD, matK, rbcL, ndhF, rpoC2, ycf2, and ycf1 (Table S1). These selected sites were highly concentrated in a few genes, with ycf1 harboring the most (22 sites), followed by ycf2 (7), rpoC2 (7), and ndhF (6), while the remaining genes each contained one to three selected codons.
This enrichment in ycf1 and ycf2 is consistent with patterns observed across angiosperms. In Zingiber, for example, these genes exhibited 52 and 24 positively selected sites, respectively (Jiang et al. 2023), and similar selection signatures have been reported in Erigeron (Asteroideae, Asteraceae) (Abdullah, Rahmatulla, et al. 2025). The multiple selected codons detected in ndhF—a gene frequently implicated in adaptive radiations—mirror findings in Erigeron and other angiosperm lineages (Abdullah, Rahmatulla, et al. 2025; Corvalán et al. 2023). Concurrent positive selection in ndhF and rpoC2, which encodes a core subunit of the plastid‐encoded RNA polymerase involved in chloroplast transcription, has also been documented in Oryza, potentially reflecting adaptation to diverse ecological or light conditions (Gao et al. 2019).
The selection landscape in V. encelioides broadly parallels patterns documented throughout Asteraceae (Table S1), where adaptive evolution consistently targets large open reading frames (ycf1, ycf2) and genes central to photosynthesis or transcriptional regulation. The relatively high density of selected codons in ycf1 and ndhF observed in V. encelioides is characteristic of these genes across the family rather than unique to this species. Thus, while these molecular signatures reflect conserved evolutionary pressures common to Asteraceae, they do not directly account for the species' invasive potential. This suggests that invasiveness in V. encelioides may instead be linked to genes in the nuclear genome.
Nucleotide Polymorphism and Phylogenetic Analysis
3.5
Nucleotide diversity (Pi) across CDS, tRNA, rRNA, intronic, and IGS regions was calculated from an alignment of the two Verbesina cp genomes. IGS regions exhibited the highest nucleotide diversity (Pi = 0.0093), followed by introns (Pi = 0.0043) and CDS (Pi = 0.0030) (Figure 6). Among CDS genes, psbF, rps15, psbT, ycf1, and psbM showed the highest variation (0.00952–0.01667). Highly polymorphic IGS regions included trnD‐trnY, atpA‐trnR, rpl32‐trnL, ccsA‐ndhD, and trnL‐ccsA (0.03774–0.14729) (Table 4). trnL‐ccsA had the highest missing data (17.8%), whereas trnD‐trnY showed the highest nucleotide polymorphism (0.77% missing).
Nucleotide diversity of chloroplast genome regions. (A) Coding regions, including protein‐coding genes, tRNA genes, and rRNA genes. (B) Non‐coding regions, including intergenic spacer regions and intronic regions.
Elevated diversity in IGS regions reflects reduced functional constraints, permitting neutral mutations to accumulate. Highly variable CDS genes such as ycf1 and psbM may experience relaxed selective pressure or lineage‐specific adaptation. These polymorphic regions differ from those previously reported in Blumea and Artemisia (Iram et al. 2019; Abdullah et al. 2021), underscoring the value of species‐specific markers for phylogenetic resolution and DNA barcoding (Ahmed et al. 2013; X. Li et al. 2015; H. Li et al. 2025).
The phylogenetic tree (Figure 7) robustly illustrates the evolutionary relationships among the major clades, with most key nodes receiving high bootstrap support (≥ 99%), indicating strong confidence in the topology. However, some nodes showed relatively lower support (bootstrap values of 62, 74, and 81). This reduced support may reflect rapid divergence during the evolutionary history of these lineages, limiting the resolution power of molecular markers, or may indicate reticulate evolution, a phenomenon previously documented in subfamily Asteroideae (Abdullah, Mehmood, et al. 2020). Nevertheless, the monophyly of major clades remains well‐supported and consistent with previous reports (Schilling and Panero 2011; Moraes and Panero 2016; Moreira et al. 2023).
Maximum likelihood phylogenetic inference among Heliantheae. All nodes with bootstrapping values ≤ 99% are shown. The V. encelioides , which is sequenced in the current study, has been shown in bold.
Within Heliantheae, species formed a distinct clade, with V. encelioides grouping with V. alternifolia (bootstrap 100%), confirming Verbesina monophyly. The Verbesina clade was sister to Silphium and Echinacea, while Bidens, Cosmos, and Coreopsis occupied more basal positions. These relationships align with previous plastid‐ and nuclear‐based phylogenetic analyses (Schilling and Panero 2011; Moraes and Panero 2016; Moreira et al. 2023), demonstrating the utility of cp genomes for resolving relationships in taxonomically challenging groups.
Conclusion
4
This study reports the first complete cp genome of Verbesina encelioides , which shows structural conservation consistent with other Asteraceae species. Comparative analyses revealed high similarity with V. alternifolia , with greater variability in intergenic spacers and introns, and several hypervariable regions and SSRs that provide useful markers for population and biogeographical studies. Positive selection was detected at 62 codons across 19 genes, reflecting patterns common in Asteraceae rather than traits linked to invasiveness, suggesting that invasive features are more likely encoded in the nuclear genome. Phylogenetic results confirmed the monophyly of Verbesina within Heliantheae.
These genomic resources offer practical value for conservation genetics, evolutionary research, and future assessments of the species' invasive dynamics.
Author Contributions
Rushan Yan: conceptualization (equal), data curation (equal), writing – original draft (equal). Abdullah: conceptualization (equal), formal analysis (equal), resources (equal), writing – review and editing (equal). Jingjing Jia: data curation (equal), investigation (equal). Madiha Islam: investigation (equal), methodology (equal). Hui Li: investigation (equal), methodology (equal). Mengyang Liu: data curation (equal), formal analysis (equal). Bartholomew Yir‐erong: conceptualization (equal), formal analysis (equal), investigation (equal), validation (equal), writing – review and editing (equal). Xiaoxuan Tian: conceptualization (equal), visualization (equal), writing – review and editing (equal).
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 82474031).
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Table S1: Analysis of positive selection.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abdullah , A. Fatima , C. Chen , et al. 2025. “The Chloroplast Genome of Trollius acaulis: Insights Into Comparative Genomics and Phylogenetic Relationships.” Journal of Asia‐Pacific Biodiversity: S 2287884 X 2500038 X. 10.1016/j.japb.2025.03.008. · doi ↗
- 2Abdullah , D. Haram , R. Yan , et al. 2025. “Inverted Repeats Dynamics Shape Asclepiadoideae (Apocynaceae) Chloroplast Genomes: Effects on Genome Size, Gene Content, Structural Arrangement, and Mutation Rate.” BMC Genomics 26, no. 1: 697. 10.1186/s 12864-025-11839-9.40721999 PMC 12302592 · doi ↗ · pubmed ↗
- 3Abdullah , C. L. Henriquez , F. Mehmood , et al. 2020. “Complete Chloroplast Genomes of Anthurium huixtlense and Pothos scandens (Pothoideae, Araceae): Unique Inverted Repeat Expansion and Contraction Affect Rate of Evolution.” Journal of Molecular Evolution 88, no. 7: 562–574. 10.1007/s 00239-020-09958-w.32642873 PMC 7445159 · doi ↗ · pubmed ↗
- 4Abdullah , H. Li , R. Yan , et al. 2025. “Evolutionary Dynamics of the Chloroplast Genome in Daphne (Thymelaeaceae): Comparative Analysis With Related Genera and Insights Into Phylogenetics.” FEBS Open Bio: 2211‐5463.70143. 10.1002/2211-5463.70143.PMC 1295575541098041 · doi ↗ · pubmed ↗
- 5Abdullah , F. Mehmood , A. Rahim , P. Heidari , I. Ahmed , and P. Poczai . 2021. “Comparative Plastome Analysis of Blumea, With Implications for Genome Evolution and Phylogeny of Asteroideae.” Ecology and Evolution 11, no. 12: 7810–7826. 10.1002/ece 3.7614.34188853 PMC 8216946 · doi ↗ · pubmed ↗
- 6Abdullah , F. Mehmood , I. Shahzadi , et al. 2020. “Chloroplast Genome of Hibiscus rosa‐sinensis (Malvaceae): Comparative Analyses and Identification of Mutational Hotspots.” Genomics 112, no. 1: 581–591. 10.1016/j.ygeno.2019.04.010.30998967 · doi ↗ · pubmed ↗
- 7Abdullah , A. Rahmatulla , C. Chen , et al. 2025. “Comparative Chloroplast Genomics of Erigeron (Asteroideae, Asteraceae).” BMC Plant Biology 25, no. 1: 1356. 10.1186/s 12870-025-07484-9.41068593 PMC 12512410 · doi ↗ · pubmed ↗
- 8Abdullah , I. Shahzadi , F. Mehmood , et al. 2019. “Comparative Analyses of Chloroplast Genomes Among Three Firmiana Species: Identification of Mutational Hotspots and Phylogenetic Relationship With Other Species of Malvaceae.” Plant Gene 19: 100199. 10.1016/j.plgene.2019.100199. · doi ↗