From “metabolic storm” to “immune paralysis”: the dynamic evolution of macrophages and metabolism reprogramming in ARDS
Jia Tang, Mi Yan, Mengchun Li, Zhangxue Hu, Kun Zhou

TL;DR
This review explores how macrophage metabolism changes during sepsis-induced ARDS and how these changes could be targeted for treatment.
Contribution
The paper provides a comprehensive overview of macrophage metabolic reprogramming and its therapeutic implications in sepsis-associated ARDS.
Findings
Macrophage metabolic reprogramming evolves from a 'metabolic storm' to 'immune paralysis' in ARDS.
Glycolysis and oxidative phosphorylation influence macrophage polarization and lung tissue damage.
BMMSCs and MDSCs offer potential therapeutic strategies by altering macrophage metabolism.
Abstract
Sepsis is characterized by high mortality and a complicated pathological mechanism. Macrophages play a crucial role in the initiation and progression of sepsis-associated acute respiratory distress syndrome (ARDS). Macrophage functional states and polarization phenotype have been significantly influenced by metabolic programming. This review delineates the metabolic reprogramming of macrophages from the initial ‘metabolic storm’ to subsequent ‘immune paralysis’ in sepsis-associated ARDS. It focuses on the interplay between macrophage polarization (classical activated macrophages (M1) and alternative activated macrophages (M2) phenotypes) and key metabolic pathways, including glycolysis and oxidative phosphorylation (OXPHOS). Furthermore, it explains the molecular mechanism underlying the metabolic pattern’s influence on macrophage and lung tissue damage. The final section of this review…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
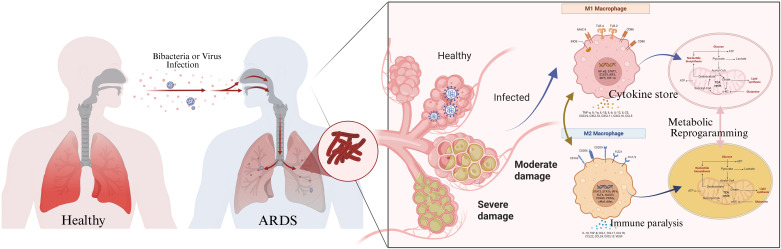 Figure 1
Figure 1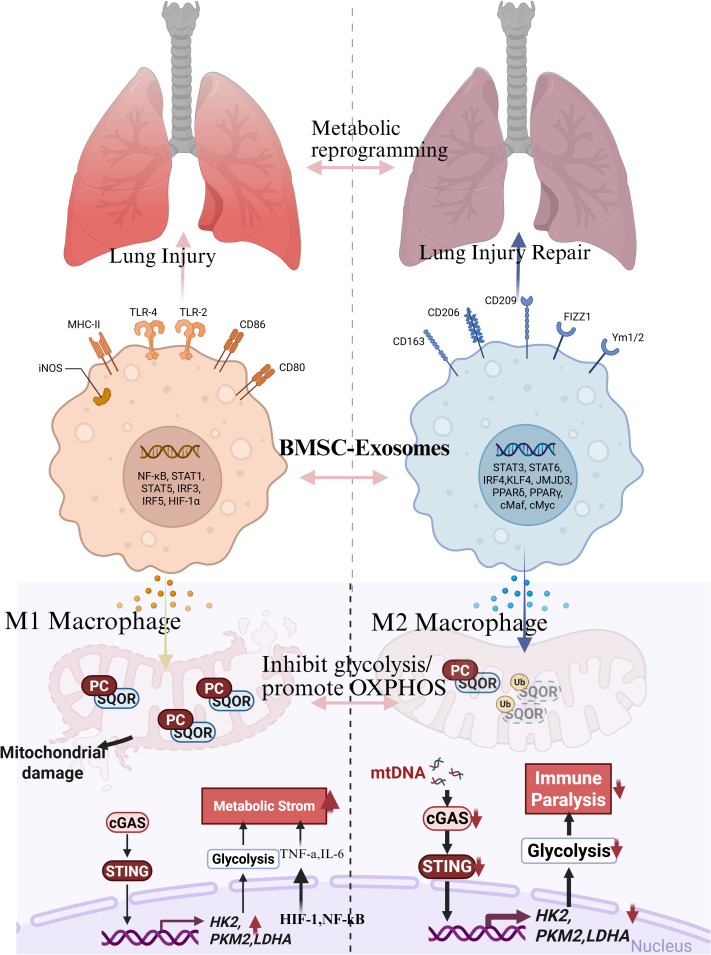 Figure 2
Figure 2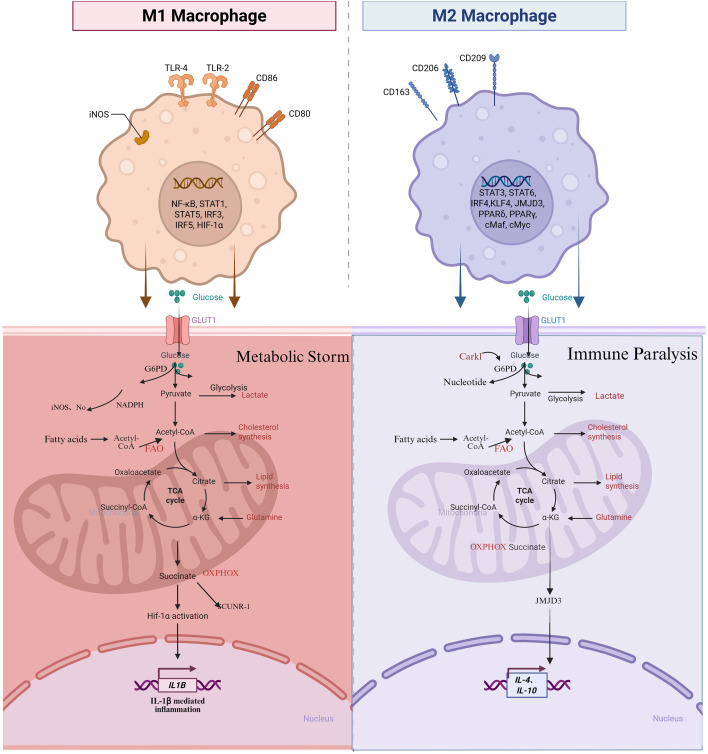 Figure 3
Figure 3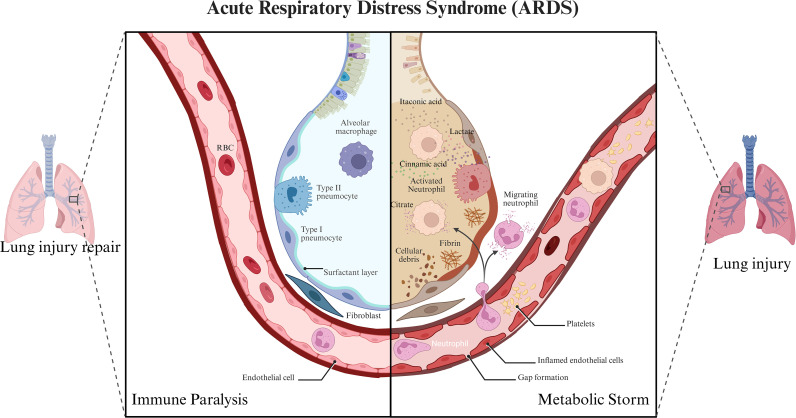 Figure 4
Figure 4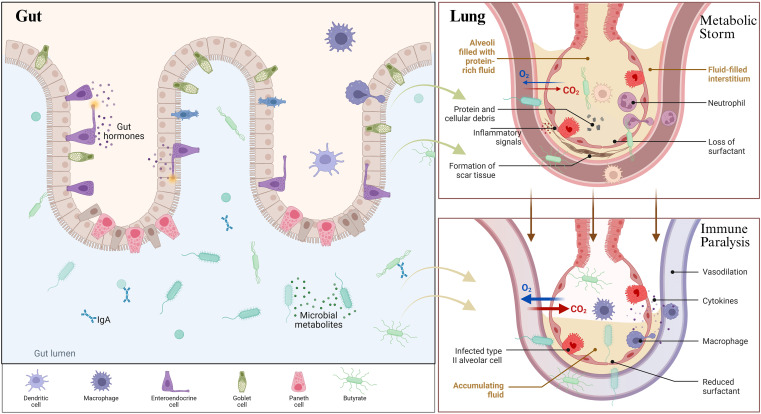 Figure 5
Figure 5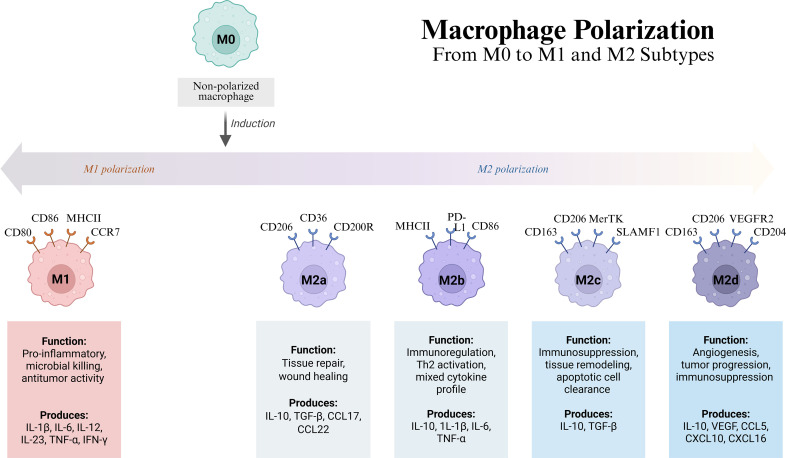 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsImmune cells in cancer · Immune Response and Inflammation · Immune responses and vaccinations
Introduction
1
Sepsis is a life-threatening syndromic entity with a critical constellation of signs and symptoms (1, 2). Sepsis results from a dysregulated host response to infection. It typically begins with an initial hyperinflammatory phase (systemic inflammatory response syndrome, SIRS), characterized by excessive pro-inflammatory cytokine release (2, 3). This can subsequently transition to a compensatory anti-inflammatory phase, leading to immunosuppression. ARDS caused by sepsis has a multi-faceted pathophysiology involving immune dysregulation, endothelial and alveolar epithelial damage, and altered metabolism, all of which compromise pulmonary function (3, 4).
Macrophages, as a central aspect of sepsis and ARDS immunopathology, are innate immune cells that activate an inflammatory response while simultaneously contributing to its suppression due to their plasticity and polarization states (5). Macrophages are classically polarized into M1 and M2 phenotypes, with proinflammatory and microbicidal properties in the former and anti-inflammatory and tissue-repair functions in the latter (5). ARDS disrupts explicitly the balance between M1 polarization support and M2 polarization, leading to metabolic acidosis and tissue damage in the alveoli through inflammation, and ultimately, to M2 polarization, which is associated with resolution and tissue recovery (6).
Metabolic reprogramming—a shift in cellular metabolic states—is intrinsically linked to macrophage polarization and function (7, 8). This includes glycolysis, OXPHOS, fatty acid oxidation (FAO), and amino acid metabolism pathways, which are associated with a proinflammatory phenotype and effector functions in distinct ways (9, 10). The Warburg effect, which links aerobic glycolysis with energy demands and proinflammatory mediator production in M1 macrophages, contrasts with OXPHOS and FAO, used to sustain anti-inflammatory functions in M2 macrophages. Metabolic reprogramming of pulmonary macrophages is a key factor that integrates bioenergetics and immune responses, influencing the progression of sepsis-induced ARDS (11, 12), which is regulated by signaling pathways and epigenetic mechanisms. Hypoxia-inducible factor (HIF-1α), a major regulator of cellular adaptation during hypoxia and metabolic stress, is involved in enhancing glycolysis and M1 macrophage polarization, which exacerbates lung inflammation (10). Other key signaling pathways include sirtuin 6 (SIRT6), which promotes anti-inflammatory macrophage polarization and autophagy in septic lungs, and ankyrin demonstrated domain 22 (ANKRD22), which enhances M1 polarization via interferon regulatory factor 3 (IRF3) in sepsis-induced ARDS (13). Non-coding RNAs and circular RNAs mediate macrophage polarization through competing endogenous RNA (ceRNA) mechanisms that regulate the inflammatory microenvironment of sepsis-ARDS (14).
“Metabolic storm” is characterized by increased glycolytic flux, reactive oxygen species (ROS) production, and M1 macrophage-driven release of proinflammatory cytokines. This is followed by “immune paralysis,” or an immunosuppressed state, in which metabolic changes, metabolic exhaustion, and M2 polarization create a barrier to pathogen clearance, thereby increasing the risk of nosocomial infections. This immune suppression, known as immunosuppression or exhaustion, underscores the need for M2 to balance M1 and M2 in defending the host and repairing tissue without causing harm.
BMMSCs have been utilized to minimize glycolysis while promoting M2 polarization of alveolar macrophages (15, 16). Furthermore, it explains the molecular mechanism underlying the metabolic pattern’s influence on macrophage and lung tissue damage. The final section of this review focuses on the therapeutic implications of BMMSCs and MDSCs, which alter macrophage metabolic reprogramming. Conceptualizing the molecular and metabolic mechanisms that govern macrophage function in sepsis-ARDS as integrated network management system (INMS) provides a critical framework for elucidating pathogenesis and identifying targeted interventions. This review systematically examines the landscape alterations that occur in sepsis-ARDS, with a particular focus on macrophage metabolic reprogramming and its dynamic impact on immune modulation and clinical therapeutic implications (Figure 1).
The dynamic evolution of macrophages and metabolic reprogramming in sepsis-associated ARDS. (Created with Biorender.com).
Macrophage polarization in sepsis-induced ARDS
2
Dynamics of macrophage polarization in sepsis-induced ARDS
2.1
M1 macrophages in sepsis-induced ARDS: pro-inflammatory roles and mechanisms
2.1.1
M1 macrophages, also known as classically activated macrophages, are crucial in the early inflammatory response because they secrete large amounts of proinflammatory cytokines, including tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β). Macrophage polarization to the M1 phenotype is associated with increased glycolysis and the expression of proinflammatory genes, which continuously generate inflammatory signals. Apolipoprotein B mRNA editing enzyme catalytic subunit 3A (APOBEC3A), an RNA editing enzyme, encourages M1 polarization by targeting cytokine gene expression and promoting inflammatory cytokine secretion (17). This excessive proliferation of the proinflammatory response is evident in the “metabolic storm” phase of sepsis-induced ARDS, during which metabolic repolarization drives macrophage overactivation.
M1 macrophages also produce ROS and pro-oxidant enzymes, such as nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase and cyclooxygenase-2, which contribute to increased oxidative stress and lipid peroxidation, they may further exacerbate tissue injury. Although some natural compounds such as carnosine can reverse these pro-oxidant and proinflammatory responses through upregulating antioxidant enzyme expression and reducing IL-1β levels, opportunities exist for novel therapeutic strategies to suppress M1 macrophage-induced inflammation Also, the inflammatory behavior of M1 macrophages is not fixed; indeed, interleukin (IL)-21, acts to impair M1 inflammatory capabilities, meaning that the cytokine environment plays a crucial role in determining M1 macrophage performance (18, 19). In pathological conditions, such as sepsis and ARDS, M1 macrophages contribute to alveolar barrier disruption by secreting TNF-α and IL-1β, which increase vascular permeability and promote leukocyte recruitment. This is exemplified by studies showing that vascular endothelial growth factor-A (VEGFA), which is upregulated in ARDS, drives M1 polarization and the concomitant release of proinflammatory cytokines, thereby exacerbating lung injury (20).
M1 macrophages produce high levels of hydrogen peroxide and other ROS, which sustain oxidative stress and tissue damage in inflamed lungs by collaborating with neutrophil-derived myeloperoxidase (21). The metabolic reprogramming that facilitates the proinflammatory phenotype of M1 macrophages is the adaptation of glycolysis. This is evidenced in the regulation of glucose transporter one and inflammatory mediators associated with M1 polarization (22). Inflamed tissues are hypoxic, which further increases M1 polarization through c-Jun N-terminal kinase (JNK)/p65 signaling pathways (23). The pathological role of M1 macrophages is observed in chronic inflammatory diseases and autoimmune conditions, all of which share tissue damage and fibrosis. Sulforaphene is a therapeutic agent that targets the NOD-like receptor (NLR) inflammasome, specifically the NLRP3 inflammasome, thereby inhibiting M1 polarization and inflammation (24).
M2 macrophages in sepsis-induced ARDS: anti-inflammatory and reparative functions
2.1.2
M2 Macrophages are of paramount importance due to their role in inflammation suppression and tissue repair. In this regard, this type is instrumental in sepsis-induced ARDS, in which a person’s immune response transitions from hyperactivity to immune suppression. The type secretes anti-inflammatory cytokines, such as interleukin-10 (IL-10) and transforming growth factor beta, which are instrumental in reducing inflammation. M2 macrophage is not homogeneous. Instead, it comprises several subtypes with distinct functions, almost all of which are linked to immunosuppression and repair. M2a and M2c subtypes primarily mediate the anti-inflammatory response and tissue regeneration. M2b reservoirs possess both proinflammatory and immunoregulatory characteristics. The switch to the M2 phenotype is supported by various metabolic repurposing and signaling pathways, including FAO and respiration, that maintain their repair functions (25–28). The adverse effects of abundant M2 polarization in vivo are worsened during the sepsis immunosuppressive phase.
Research indicates that macrophage metabolism in sepsis displays distinct mixed characteristics, encompassing both the enhanced glycolysis typical of M1 macrophages and the OXPHOS metabolic components characteristic of M2 macrophages. This metabolic cross-regulation not only reflects macrophages’ adaptive adjustments to energy metabolism within the complex microenvironment of sepsis but also underscores their functional diversity and heterogeneity (29). During the immune response to sepsis, macrophages demonstrate a high degree of plasticity and heterogeneity in their metabolic state and function. The synergistic activation of dual signaling molecules plays a pivotal role in modulating inflammatory metabolic pathways. High mobility group box 1 protein (HMGB1) and cold-inducible RNA-binding protein (CIRP), as unique signaling molecules in sepsis, jointly activate the toll-like receptor 4 (TLR4)/nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and HIF-1a signaling pathways within macrophages, thereby enhancing both glycolytic metabolism and the pro-inflammatory response. Furthermore, these two signaling molecules also activate HIF-1α, a transcription factor that is central to the metabolic reprogramming of macrophages. It promotes the expression of key glycolytic enzymes, boosts the activity of the glycolytic pathway, and provides a metabolic foundation for rapid energy provision and the synthesis of inflammatory mediators in macrophages (30).
M1/M2 polarization imbalance and ARDS progression
2.1.3
The M1 and M2 polarization types in macrophages, as previously described, are key factors in the development and progression of ARDS. The balance between these two types mainly governs the inflammatory phase, tissue repair, and clearance phase. In ARDS patients, lung macrophages show a higher M1/M2 ratio due to the overexpression of secreted phosphoprotein 1 (SPP1). This gene increases the ratio by inhibiting HIF-1α, thereby exacerbating lung injury and fibrosis in ARDS models (31, 32). Furthermore, DAMPs can exacerbate inflammation by disrupting the member of the Ras oncogene family (Rab26) maturation following erythropoietin receptor (EPOR) activation and suppressing M2 polarization, thereby prolonging the inflammatory response (33) (Figure 2).
From “metabolic storm” to “immune paralysis”: the dynamic evolution of macrophages and metabolic reprogramming in sepsis-associated ARDS. (Created with Biorender.com).
Otherwise, therapeutic interventions targeting macrophage polarization have illustrated promise. As previously discussed, mesenchymal stem cells (MSCs) and their exosomes can initiate M2 polarization by inhibiting glycolysis, thereby improving outcomes in ARDS (34). Additionally, pharmacological agents such as rhein and luteolin have been demonstrated to promote M2 polarization (35, 36). Similarly, CD274 has been reported to act as a negative regulator of M1 polarization while promoting an M2 phenotype, suggesting a potential new diagnostic or therapeutic approach (37). Furthermore, due to its dynamic plasticity, metabolic reprogramming is another key biological feature, with FAO and glycolysis influencing the macrophages’ phenotype and function, especially in the context of ARDS (38–41) reported that the neuropeptide calcitonin gene-related peptide partially corrected the imbalance of M1/M2 in bronchoalveolar lavage fluid from ARDS patients. Sepsis is characterized by a high degree of heterogeneity in its pathological features, particularly in terms of the metabolic reprogramming and functional manifestations of macrophages.
Molecular mechanisms of macrophage metabolic reprogramming
2.2
Enhanced glycolysis drives M1 polarization
2.2.1
Proinflammatory M1 macrophages heavily rely on enhanced glycolysis to rapidly generate energy for synthesizing and secreting inflammatory mediators. This metabolic reprogramming supports their effector functions during acute inflammatory responses. When activated by stimuli such as lipopolysaccharide (LPS) and interferon-gamma (IFN-γ), macrophages shift from OXPHOS to aerobic glycolysis, known as the Warburg effect, to meet the increased bioenergetic and biosynthetic demands of inflammation. This glycolytic shift enables faster adenosine triphosphate (ATP) production, albeit less efficiently, allowing rapid responses to pathogens or tissue injury (42). Key glycolytic enzymes, including hexokinase 2 (HK2), pyruvate kinase M2 (PKM2), phosphofructokinase-2/fructose-2,6-bisphosphatase 3 (PFKFB3), and lactate dehydrogenase A (LDHA), are upregulated during M1 polarization, enhancing glucose uptake and lactate production (43, 44). Following these metabolic reprogramming events, the transcription factor HIF-1α plays a vital role in regulating cellular processes. HIF-1α activates glycolytic gene expression. Under inflammatory conditions, HIF-1α is stabilized in a tumor, even under normoxia, through signaling pathways such as the Janus kinase 2 (JAK2)/signal transducer and activator of transcription 1 (STAT1) axis, activated by IFN-γ, or the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and mTORC1 pathways (45). The JAK2/STAT1 signaling pathway induced by IFN-γ promotes PFKFB3 expression, thereby boosting Glycolysis and M1 polarization in vascular inflammation. Similarly, the JNK/Cyclooxygenase-2 (COX-2)/HIF-1α axis enhances glycolysis in the M1 polarization of human immunodeficiency virus (HIV)-1-infected macrophages, linking metabolic reprogramming to chronic inflammation (46). This metabolic pathway not only supplies energy but also produces intermediates that act as signaling molecules to upregulate inflammatory genes. Nitric oxide (NO) produced by inducible nitric oxide synthase (iNOS) stabilizes HIF-1α, promoting glycolysis and inflammatory cytokine production (47). Additionally, pyruvate kinase M2 is a key regulator of glycolysis and inflammatory gene expression in M1 macrophages, switching between its tetrameric enzymatic form and dimeric transcriptional coactivator form (48). Several endogenous and exogenous factors influence this glycolytic reprogramming. RNA-binding motif protein 4 (RBM4) inhibits M1 polarization by suppressing STAT1-mediated glycolysis (49). Natural compounds such as Cassiaside C and berberine decrease M1 polarization by inhibiting glycolysis through HIF-1α and mTORC1 signaling, respectively, and by activating AMP-activated protein kinase (AMPK) (50). Factors like C-X-C motif chemokine receptor 1 (CXCR4) support M1 polarization by promoting glycolysis via the phosphatidylinositol-3 kinase (PI3K)/protein kinase B (AKT)/mTOR pathway. Additionally, metabolic enzymes influence macrophage polarization through epigenetic modifications that do not require transcription. Hexokinase 2 (HK2)-driven glycolysis induces lactate signaling, leading to histone lactylation, which further reinforces M1 gene expression in liver macrophages during metabolic disease (51).
OXPHOS and FAO support M2 polarization
2.2.2
M2 macrophages, which possess the metabolic characteristics of anti-inflammation and tissue repair, depend heavily on OXPHOS and FAO to meet energy requirements and sustain their functional phenotype. This metabolic characteristic is the opposite of M1 macrophages, which are glycolytic-dependent and support proinflammatory responses. The OXPHOS and FAO pathways enable M2 macrophages to function as anti-inflammatory cells, promoting the resolution of inflammation.
Several studies have clarified the role of fatty acid metabolism in promoting M2 polarization. The fatty acid-binding protein 5 (FABP5) has been identified as a key regulator in macrophage alternative activation. Deletion of FABP5 in myeloid cells results in an accumulation of free long-chain unsaturated fatty acids such as oleic acid, which enhances fatty acid β-oxidation, the tricarboxylic acid (TCA) cycle, and OXPHOS, thereby encouraging M2 polarization through activation of the peroxisome proliferator-activated receptor gamma (PPARγ) signaling pathway (52). This highlights the essential role of lipid handling and mitochondrial metabolism in determining macrophage phenotypes. Similarly, in lung inflammation models, therapeutic interventions have been illustrated to suppress proinflammatory cytokines and promote M2 polarization by modulating fatty acid metabolism via the Akt signaling pathway. Treatment reduces fatty acid synthesis (FAS) and boosts FAO, leading to increased expression of M2 markers such as arginase-1 (Arg-1) and decreased iNOS expression (53). These metabolic shifts are mediated by downstream targets of Akt signaling, including ATP-citrate lyase (ACLY) and peroxisome proliferator-activated (PPARa), which regulate FAS and oxidation, respectively.
Several essential pathways regulate transcriptional metabolic reprogramming in M2 macrophages. Activation of PPAR family members, especially PPARγ and PPARα, triggers FAO and mitochondrial biogenesis gene programs. Additionally, signaling pathways such as the AMPK pathway enhance mitochondrial oxidative metabolism and FAO, further supporting M2 polarization (54). Other metabolic pathways involving mTOR and HIF-1α in target macrophages include the mTOR and HIF-1α pathways, which balance glycolysis and oxidative metabolism in response to environmental signals (55).
Additionally, mitochondrial chaperones, including heat shock protein 60, have been reported to maintain mitochondrial function and promote FAO in macrophages. The high expression of Heat Shock Protein 60 (HSP60) leads to FAO. Meanwhile, the activity of MRC complexes remains unchanged and is associated with low inflammation and restored metabolic health in disease models, including nonalcoholic fatty liver disease (NAFLD) (56). These studies conclude that mitochondrial quality control networks support the oxidative metabolic mode underlying M2 macrophage function, which is essential. M2 macrophages rely on oxidative metabolism in FAO and OXPHOS, facilitating tissue repair (Figure 3) (57, 58).
M1/M2 polarization imbalance and macrophage metabolic reprogramming (42). (Created with Biorender.com).
Regulation of macrophage function by metabolic intermediates
2.2.3
Metabolic intermediates like lactate and citrate dynamically influence macrophage polarization and function by modulating inflammatory signaling pathways (Figure 4). First, lactate, which was traditionally seen as a metabolic byproduct of glycolysis, is now recognized as a powerful signaling molecule that determines macrophage phenotype. Adipocyte-secreted lactate acts as a danger signal. It promotes M1-like macrophage polarization by directly binding to prolyl hydroxylase domain-containing protein 2, thereby stabilizing HIF-1α and upregulating its downstream target, IL-1β, which contributes to systemic and adipose tissue inflammation (59, 60).
Regulation of macrophage function by metabolic intermediates in ARDS. (Created with Biorender.com).
Citrate, a critical metabolite of the Tricarboxylic acid cycle (TAC), supports the biosynthesis of inflammatory mediators. The accumulation of citrate enhances prostaglandin and NO biosynthesis by activated macrophages, thereby increasing inflammatory signaling (61–63). Similarly, since succinate binds to succinate receptor 1 (SUCNR1) to act as a signaling metabolite, it influences macrophage development by inducing M2 hyperpolarization via the Gq signaling pathway (64). However, succinate-dependent macro-specific activity is context-dependent, with evidence supporting both enhanced anti-inflammatory and proinflammatory activity upon succinate binding, respectively (65)(Figure 4).
Citrate and lactate also intersect with lipid metabolism pathways to regulate macrophage polarization. Arachidonic acid metabolism, downstream of citrate, controls macrophage alternative activation by modulating OXPHOS via PPARγ, with prostaglandin E2 (PGE2) derived from arachidonic acid facilitating M2 polarization (66). Moreover, lipid metabolites such as leukotriene B4 (LTB4), produced via Arachidonate 5-lipoxygenase (ALOX5) from arachidonic acid, recruit M2 macrophages by activating the PI3K pathway, promoting tumor progression in intrahepatic cholangiocarcinoma (67). These findings highlight the complex interplay between metabolic intermediates and lipid-derived metabolites in shaping macrophage phenotypes.
Cinnamic acid facilitates the polarization of anti-inflammatory M2 macrophages, a process intricately linked to the activation of the STAT6 and PPARγ signaling pathways (68). Additionally, cinnamic acid may modulate the functional status and polarization trajectory of macrophages in sepsis by intervening in signaling pathways such as extracellular regulated protein kinases (ERK)1/2 (69). This metabolic reprogramming serves as the foundation for the transition of macrophage polarization states. Moreover, cis-aconitate can also orchestrate the shift in macrophage polarization states by adjusting histone acetylation levels and consequently impacting gene expression patterns.
Itaconic acid promotes antioxidant response by activating Nrf2 signaling pathway, further exerting its immune regulatory function. Nrf2 is the main antioxidant transcription factor in cells, capable of inducing the expression of various antioxidant enzymes and protecting cells from oxidative stress damage. Itaconic acid promotes the stabilization and nuclear translocation of Nrf2 through covalent modification with Keap1, enhancing glutathione synthesis, inhibiting ROS generation, reducing inflammatory factor release, and improving sepsis-induced tissue damage (70). In sepsis models, the accumulation of succinic acid and its induced HIF-1α stabilization are considered important factors in triggering excessive inflammatory responses (71). Through this mechanism, succinic acid promotes the expression of pro-inflammatory factors and the inflammatory cascade, leading to aggravated tissue damage. In sepsis and related inflammatory states, succinic acid promotes the chemotaxis and activation of macrophages through succinate receptor 1 (GPR91), exacerbating the inflammatory response (64).
In addition to lactate and citrate, other metabolites such as butyrate — a short-chain fatty acid generated by gut microbiota shape macrophage polarization by stimulating the M2 phenotypes and quench the production of proinflammatory cytokines(Figure 5). Moreover, microbial metabolites, including reuterin, instruct tumor-associated macrophages to adopt an M1 antitumor phenotype through a reprogramming mechanism, thereby enhancing the efficacy of immunotherapy.
Regulation of macrophage function by gut metabolic intermediates. (Created with Biorender.com).
These metabolites modulate macrophage function through various signaling pathways, including stabilizing transcription factors, activating or inhibiting nuclear receptors, regulating kinase cascades, and inducing epigenetic modifications that influence gene expression. Lactate competitively inhibits α-ketoglutarate binding to prolyl hydroxylase (PHD2), leading to stabilization of HIF-1α and activation of proinflammatory macrophages. Itaconate exerts an anti-inflammatory effect by modulating the Kelch-like Epichlorohydrin (ECH)-ECH-associated protein (Keap1)-Nrf2 pathway and inhibiting the stimulator of interferon genes (STING) pathway, thereby reprogramming macrophage immune responses and metabolism (72).
Functional characteristics of macrophages in the “metabolic storm” phase
2.3
Metabolic activity and enhanced inflammatory response
2.3.1
Within hours to days after the onset of sepsis, the condition typically progresses to a “metabolic storm” phase, marked by hyperactive immune cell metabolism and the release of a substantial amount of inflammatory factors, alongside systemic inflammatory response syndrome. During this stage, the host’s recognition of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) activates the innate immune system, setting off a widespread and unrestrained immune response that culminates in a cytokine storm. This cytokine storm is characterized not only by the explosive release of pro-inflammatory cytokines such as TNF-α and IL-6 but also by aberrant metabolic changes, particularly the metabolic reprogramming of immune cells. M1 macrophages, a classic example of pro-inflammatory cells, exhibit metabolic reprogramming primarily characterized by enhanced glycolytic activity and disruption of the TCA cycle, accompanied by the accumulation of key metabolites like succinate and citrate. These metabolites not only function as intermediates in energy and material metabolism but also play a pivotal role by facilitating inflammatory signal transduction. Specifically, activated M1 macrophages demonstrate significantly elevated glycolysis, primarily driven by the upregulation of the glucose transporter 1 (GLUT1), key enzymes such as hexokinase 2, PFKFB3, and the glycolysis regulatory factor HIF-1α. These molecules collectively enhance glucose uptake and metabolism, thereby meeting the rapid energy demands of macrophages and facilitating the synthesis of inflammatory mediators (73). During the metabolic storm phase, M2 macrophages exhibit low-level expression of their markers, characterized by reduced expression of Arg-1band CD206, along with decreased activity of enzymes involved in FAO. Typically, M2 macrophages perform anti-inflammatory and tissue-repairing functions, and their activity hinges on the activation of metabolic pathways such as FAO.
Furthermore, the synthesis of other metabolic intermediates, such as succinate, might stabilize HIF-1α, a potent inducer of glycolysis and inflammatory cytokine production, thereby increasing its localization and further activating the inflammatory loop (73). This metabolic crisis in macrophages leads to progressive tissue damage by fueling inflammation and hindering resolution. Sequestered mitochondria and accumulated ROS, inherent in sepsis, are produced by macrophages and present a detailed pattern of defective clearance of dysregulated cells and Alveolar destruction. These conditions are correlated with poor outcomes in ARDS patients (74). Increased glycolysis and inflammasome-dependent upregulation of inflammasome NLRP3, involved in the maturation and release of IL-1β, an essential cytokine of sepsis-induced inflammation, are enhanced by mitochondrial inhibition (75–78).
Activation of metabolic regulatory networks
2.3.2
Metabolic regulatory networks in macrophages during sepsis-induced ARDS are orchestrated by a complex interplay between signaling pathways and metabolic enzymes, ultimately driving the inflammatory response and macrophage polarization. HIF-1α promotes the transcription of glycolytic enzymes and glucose transporters, thus implementing a metabolic switch from OXPHOS to aerobic glycolysis that supports the energetic and biosynthetic requirements of classically activated M1-polarized macrophages. The findings from experimental models demonstrated that HIF-1α knockout or inactivation reduces glycolytic flux, inhibits the production of proinflammatory cytokines, and diminishes lung lesions in LPS-induced ARDS, thereby highlighting the importance of HIF-1α in aligning metabolism with inflammation (12, 79).
Furthermore, several metabolic enzymes act as key effectors downstream of these signaling pathways. Lactate dehydrogenase A, which converts pyruvate into lactate, is one of the enzymes overexpressed during glycolytic activity in inflammatory macrophages. LDHA is phosphorylated and activated, promoting NF-κB signaling through increased ROS production in a positive feedback loop that sustains inflammation in sepsis-induced ARDS lung epithelial cells (80). Kaempferol can reduce septic ARDS inflammation by inhibiting the HIF-1α, NF-κB, and PI3K-Akt pathways in alveolar macrophages, thereby decreasing inflammation and apoptosis. The interconnected relationship between inflammation and metabolic signaling may alter both pathways, potentially offering a strategy to redirect macrophage-driven inflammation in sepsis-induced lung injury.
Dysregulation of glucocorticoid metabolism in alveolar macrophages, especially reduced activity of 11β-hydroxysteroid dehydrogenase type 1 reductase (HSD-1), has been linked to increased inflammation by diminishing the anti-inflammatory effects of local glucocorticoids. Impaired HSD-1 correlates with heightened NF-κB activation and poor clinical outcomes in sepsis-induced ARDS, underscoring the significance of metabolic enzyme activity in both pro- and anti-inflammatory signaling. Recent research also highlights the role of aquaporins, particularly aquaglyceroporins like aquaporin 3 (AQP3) and aquaporin 9 (AQP9), in transporting metabolic substrates, such as glycerol, which stimulates glycolysis and energy production in immune cells. AQPs also influence key inflammatory pathways, including NF-κB and toll-like receptor 4 (TLR4), as well as the functions of macrophages and neutrophils during sepsis (81). Pharmacological targeting of AQPs has demonstrated promising results in sepsis models by reducing inflammation and organ damage, further emphasizing the role of metabolism within the immune system.
Contribution of metabolic storm to lung tissue injury
2.3.3
Metabolic storm, characterized by profound metabolic reprogramming and dysregulation, plays a pivotal role in exacerbating lung tissue injury during sepsis-associated ARDS (82, 83). Hyperglycemia promotes SARS-CoV-2 infection and monocyte activation via an HIF-1α-dependent mechanism on glycolysis, resulting in mitochondrial ROS and HIF-1α stabilization. This metabolic reprogramming not only promotes viral replication but also inhibits T cell responses and compromises epithelial cell survival, contributing to lung injury (84). Interactions with inflammatory cytokines, such as interleukin-6, exacerbate lung damage. IL-6 enhances ferroptosis in alveolar epithelial cell (AEC-II) by affecting bile acid metabolism and the farnesoid X receptor (FXR), which is critical for lung regeneration. FXR inhibition alleviates IL-6-induced ferroptosis and inflammation, as observed by cell death and 5-Ethynyl-2’-deoxyuridine (EdU) staining assays. Together, these findings highlight the crucial role of metabolic reprogramming in regulating cell death and tissue repair (85). Mitochondrial dysfunction is another hallmark of metabolic storm-induced lung injury, as demonstrated by a study of lung epithelial cells. Pallmitoylation of Cyclooxygenase-2 (COX-2) at Cys555 increases COX-2’s interaction with HK2, driving mitochondrial metabolic reprogramming and, thus, the proteolytic inflammatory injury pyroptosis via activation of the NLRP3 inflammasome in mitochondria. This same study demonstrates that palmitoylation inhibition of hexokinase 2 (HK2) at Cys555 decreases metabolic and lung injury associated with inflammation (86–88).
Simultaneously, metabolic reprogramming causes endothelial dysfunction to worsen, leading to lung injury. Growth differentiation factor 15 (GDF15) protects against sepsis-induced ALI by suppressing the HIF-1α/LDHA glycolytic axis in pulmonary ECs, thereby reducing cytokine production and leukocyte recruitment. As a result, the loss of this regulation enhances inflammation and barrier dysfunction, contributing to alveolar damage. Overall, metabolic disturbances during sepsis lead to oxidative stress, ferroptosis, pyroptosis, and apoptosis in ECs (89). Abnormalities in metabolic reprogramming—including uncontrolled glycolysis, mitochondrial dysregulation, and specific signaling pathways like HIF-1α, IGF1R, and FXR—initiate these processes. Cell injury then compromises the alveolar-capillary barrier, worsens inflammation, and impairs lung regeneration, collectively promoting tissue damage. Modulating metabolic reprogramming, including targeting glycolytic enzymes, mitochondria, and metabolic receptors, may help reduce lung injury in SAARD and improve patient outcomes (83–89).
Metabolic suppression and macrophage dysfunction in the “immunoparalysis” stage
2.4
Inhibition of energy metabolic pathways leading to immune dysfunction decline
2.4.1
On the 3rd to 7th day of sepsis progression, the immune system enters a paralysis phase, characterized by a marked reduction in the expression of pro-inflammatory factors in M1 macrophages, as evidenced by decreased secretion of TNF-α, IL-1β, and other cytokines. Concurrently, immunosuppressive molecules such as programmed death ligand 1 (PD-L1) and the anti-inflammatory cytokine IL-10 exhibit upregulated expression. This alteration in expression patterns signifies a transition of the immune system from a hyperactivated to a suppressed state, resulting in compromised host immune function and an elevated risk of infection. Additionally, the activity of glycolysis-related enzymes declines, metabolic processes slow down, and the energy supply to M1 macrophages becomes restricted, further impairing their pro-inflammatory capabilities. Multiple signaling pathways are implicated in this regulatory mechanism. Androgen receptor (AR) signaling enhances M1 polarization and pro-inflammatory factor secretion by upregulating FKBP prolyl isomerase 5 gene (FKBP5) expression and activating the NF-κB pathway, whereas AR deficiency inhibits M1 polarization and mitigates lung injury. Moreover, Nrf2 deficiency can intensify M1 polarization and inflammation, while Nrf2 plays a protective role by modulating autophagy and the PPARγ/NF-κB pathway to maintain a balance between M1 and M2 polarization (90).
Mitochondrial OXPHOS also generates much energy and ROS that kill oxygen-dependent pathogens. However, in sepsis-induced ARDS, this metabolic pluripotency appears to be pushed to the background. Studies have confirmed that glycolytic function and mitochondrial respiration are both reduced in macrophages throughout immune paralysis. This results in not only a lack of ATP supply but also cellular impairment (91).
The energy deficit results in a reduced ability for phagocytosis and impeded secretion of pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, all of which are defining features of immunosuppression. Glycolytic enzymes’ downregulation and mitochondrial dysfunction are involved in this metabolic deficiency. The inhibition of crucial glycolytic enzymes or mitochondrial complexes prevents the generation of enough energy and biosynthetic precursors that activate and support a macrophage effector response (92). Mechanistically, the suppression of metabolic pathways in macrophages during sepsis may be linked to signaling abnormalities. The latter includes low activation levels of AMP-activated protein kinase and the mammalian target of rapamycin, which maintain both immunity and cellular energy homeostasis (92).
Immunoparalysis of macrophages is further metabolically linked to tissue microenvironment-specific mitochondrial crosstalk. The harsh acidity, notably extracellular lactate or other metabolites present in the microenvironment of inflamed tissues, represses macrophage glycolysis and OXPHOS; therefore, it suppresses the function of macrophages (93). Furthermore, the overabundance of immune-assassin metabolites, including methylmalonic acid, can contribute to systemic metabolic dysfunction and exhaustion in CD8+ T cells, suggesting that metabolic disorders may have broader implications for immune activities beyond macrophages (94). Importantly, interventions targeting metabolic pathways have demonstrated promise in reversing immune dysfunction. Pharmacological activation of AMPK or inhibition of mTOR can restore metabolic activity and improve macrophage function, enhancing phagocytosis and cytokine production (93). Nanomedicine strategies to modulate metabolic processes in immune cells, such as glycolysis or mitochondrial improvements, have the potential to reverse metabolic processes associated with immune competence (95, 96).
Changes in macrophage polarization under immunosuppressive conditions
2.4.2
The increased M2 polarization phenotype observed in macrophages under immunosuppression suggests a bias toward a more anti-inflammatory, tissue-repair-oriented phenotype. Nonetheless, at the expense of an enhanced M2 orientation, the functional capacity of these macrophages is diminished, a state that skews immune regulation in an imbalanced manner, potentially promoting pathological conditions rather than ameliorating them (Figure 6). This phenomenon has been observed in the context of sepsis-associated ARDS and tumor microenvironments, and is well described in several studies. This clearly indicates that immunosuppression can negatively affect macrophage activity, regardless of the polarization state (97, 98). Furthermore, lymphatic leakage fosters an immunoregulatory environment by promoting M2 via medical irradiation, highlighting the essential role of the tissue microenvironment in macrophage polarization (99, 100).
Macrophage polarization and M2 phenotype. (Created with Biorender.com).
Anti-inflammatory cytokines, growth factors such as VEGF and PDGF, and matrix metalloproteinases secreted by immunosuppressive M2 phenotype macrophages promote tumor growth, metastasis, and immune escape. This phenotype recruits and supports other immunosuppressive cell types, such as regulatory T cells and B cells, as well as alternatively activated neutrophils, collectively forming a complex immunosuppressive niche (101–106).
Moreover, emerging evidence has highlighted the contribution of KO of macrophage polarization to metabolic reprogramming (107, 108). The M2 macrophage subtypes (M2a, M2b, M2c, M2d) exhibit diverse immunosuppressive and pro-angiogenic functions, secreting factors that promote tumor progression and modulate the activity of other immune cells to maintain an immunosuppressive microenvironment (101). Glioma stem cells secrete Follistatin-like 1(FSTL-1), which sustains stemness and induces M2 polarization via TLR2-mediated PI3K-AKT signaling, highlighting tumor-stroma crosstalk in immunosuppression (109).
Targeting metabolic enzymes and pathways holds therapeutic potential for reversing M2 polarization. Inhibition of Pim2 kinase reduces glycolytic reprogramming and M1 polarization in inflammatory arthritis. At the same time, nuclear phosphoglycerate dehydrogenase (PHGDH) regulates macrophage polarization via transcriptional repression of glutamine metabolism enzymes in breast cancer, promoting immunosuppressive M2 phenotypes (110, 111). Additionally, mitochondrial metabolism and FAO have been implicated in maintaining M2 polarization, with interventions that modulate these pathways shifting macrophages toward anti-tumor phenotypes (112–114).
Impact of immune paralysis on the prognosis of sepsis-associated ARDS
2.4.3
Immune paralysis, also known as immunosuppression, represents a major driver of adverse outcomes in ARDS associated with sepsis. In the late phase of sepsis, the previously hyperinflammatory state transitions into a state of profound immune dysfunction, characterized by the suppression of both innate and adaptive immune responses. Maladaptive changes in macrophage metabolism in sepsis contribute to a decrease in phagocytic and antigen-presenting activity, leading to immune paralysis. Whereas metabolic reprogramming involving the upregulated glycolysis and impaired mitochondrial bioenergetics promotes M1 polarization in the early phase of the septic insult, in the later course of the disease, it leads to macrophage exhaustion and reduced antimicrobial capacity, which facilitates the development of secondary infections and prolonged inflammatory-mediated lung injury (115, 116).
During the immunosuppressive phase, the amounts of pro-inflammatory cytokines produced drop, and the up-regulation, combined with the activation of NLRP3 inflammasomes, is impaired. Factors, including metabolic regulators such as HIF-1α and enzymes like Succinate Dehydrogenase (SDH), altered macrophage responses and generated a switch in their power status, affecting the pattern between immune activation and paralysis (117).
Furthermore, the immune dysfunction in immune paralysis allows recurrent infections and continuous lung tissue injury, thereby extending the disease course. The inability to properly eradicate the pathogens leads to chronic alveolar damage, increased vascular permeability, and worsened ARDS pathophysiology. There is evidence that macrophage metabolic dysregulation, such as reduced OXPHOS and increased ROS production, also induces mitochondrial damage and promotes cell death. Therefore, immune defense in the lung microenvironment becomes crippled (118, 119). This process forms the oxygen in a cycle of immune dysfunction and lung tissue injury. This highlights the need to normalize macrophage metabolism to improve the course of ARDS in sepsis.
Metabolic reprogramming of other innate immune cells, namely neutrophils and dendritic cells, during sepsis also results in an extreme immunosuppressive environment. The decrease in glycolysis and FAO in these immune cells impairs their response to infections, leading to increased cytokine production and increased susceptibility to secondary infections (120, 121). Immune paralysis brings about systemic immune depression that results in not only extension of mechanical ventilation and intensive care unit timeframe, but also high susceptibility to hospital-acquired infections, which are challenging to manage and lead to an increased possibility of a patient’s death. The recent study suggests that metabolism-based therapies can reverse the above-described process. These include agents such as metformin and itaconate derivatives, which are capable of reprogramming the macrophages’ metabolism, thereby helping to decrease excess inflammation and increase bacterial clearance capacity to alleviate lung injury in sepsis models, as well as improving the chances of survival (116). Furthermore, targeting key metabolic enzymes, such as the muscle-type 6-phosphofructokinase (PFKM), to inhibit glycolysis has demonstrated protective effects against sepsis-induced cytokine storms and organ damage (122–125).
BMSC-EXO in regulating macrophage metabolism
2.5
Immunomodulatory Functions of BMSC-EXO
2.5.1
BMSCs secrete exosomes that carry a diverse array of bioactive molecules, including microRNAs (miRNAs), proteins, and signaling lipids, which collectively contribute to their potent immunomodulatory effects. These exosomes have been demonstrated to regulate inflammatory responses and modulate immune cell functions across various disease models, underscoring their therapeutic potential. BMSC-derived exosomes (BMSC-EXO) deliver the protein JNK activating protein (JKAP) to restore the balance between pro-inflammatory Th17 cells and anti-inflammatory regulatory T cells (Tregs) by activating AKT/ERK signaling pathways, thereby alleviating disease symptoms and inflammation (126–128).
Beyond T cell modulation, BMSC-EXO also affect the functions of the innate immune cells, including macrophages and dendritic cells (129). In sepsis-induced lung injury, BMSC-EXO serum amyloid A1 (SAA1) reduces inflammation and endotoxin levels, promoting alveolar macrophage function and attenuating lung damage (130). These findings underscore the capacity of BMSC-EXO to modulate both adaptive and innate immune responses by delivering specific proteins and miRNAs that regulate key signaling pathways, including the NF-κB, NLRP3 inflammasome, and cytokine networks (131–133). These data collectively highlight the broad anti-inflammatory and immunoregulatory actions of BMSC-EXO, which are mediated through the modulation of inflammasome activation, cytokine secretion, and immune cell polarization (134). This suggests that environmental cues modulate the cargo and functional potency of BMSC-EXO, enhancing their immunoregulatory capabilities.
BMSCs-derived exosomes regulate macrophage polarization by inhibiting glycolysis
2.5.2
BMSC-EXO is rich in miRNAs, which can target and modulate key molecules within macrophages, thereby facilitating the transition from M1 to M2 polarization. Studies have revealed that miR-23a-3p, highly expressed in BMSC-EXO, can specifically target and inhibit interferon regulatory factor 1 (IRF1), a pivotal regulatory factor for M1 polarization (135). Moreover, BMSC-EXO overexpressing miR-23a-3p significantly promotes the polarization shift of macrophages from the M1 to the M2 phenotype. miR-25-3p suppresses M1 polarization and fosters M2 polarization by inhibiting the activity of the JAK2/STAT3 signaling pathway. These miRNAs orchestrate metabolic reprogramming and functional transformation in macrophages through precise modulation of inflammation-related signaling pathways (136). Additionally, BMSC-EXO carries specific proteins that also play a role in regulating macrophage polarization. Research indicates that the CISH protein delivered by BMSC-EXO can inhibit the activation of the NF-κB signaling pathway, thereby promoting macrophage polarization towards the M2 phenotype (137).
BMSC-EXO inhibits glycolysis by downregulating the expression of key glycolytic enzymes. It significantly reduces the expression of enzymes closely associated with glycolysis in macrophages, including hexokinase (HK), phosphofructokinase (PFK), and lactate dehydrogenase (LDH). Elevated activity of these enzymes drives macrophage metabolism towards glycolysis, resulting in the production of substantial amounts of lactic acid and supporting the M1 polarization phenotype. The intervention of BMSC-EXO diminishes the activity of the glycolytic metabolic pathway by lowering the stability and expression of HIF-1α, consequently reducing the expression levels of the aforementioned enzymes. This, in turn, suppresses the release of pro-inflammatory cytokines such as TNF-α and IL-6, effectively mitigating inflammatory injury and pathological alterations in lung tissue. By inhibiting the phosphorylation of p65 and p38, BMSC-EXO reduces the proportion of M1-polarized macrophages. It can also inhibit the glycolytic activity of macrophages, diminish the expression and activity of key glycolytic enzymes, and lower lactate production, thereby attenuating the metabolic characteristics of pro-inflammatory M1 macrophages. Concurrently, BMSC-EXO promotes the recovery of mitochondrial function in macrophages, elevates the level of oxidative phosphorylation, increases ATP production, and enhances the activity of metabolic pathways such as FAO, thereby facilitating the polarization of macrophages towards the M2 phenotype.
BMSCs have become potent modulators of the immune response in sepsis-induced ARDS, particularly through exosomal secretion that influences macrophage activity. A crucial mechanism by which BMSC-EXO target and cure is by inhibiting glycolysis in macrophages, thereby altering the regulatory state of these cells. Macrophages modify their metabolism during inflammation, with the M1 phenotype upregulating glycolysis through the HIF-1α pathway, thereby enhancing the regulation of glycolytic genes. This metabolic transition is expected to support the pro-inflammatory target, thereby precipitating lung damage in ARDS. Recent studies have demonstrated that exosomes secreted by BMSCs can downregulate HIF-1α expression in macrophages, leading to decreased activity of key glycolytic enzymes and a subsequent reduction in glycolytic flux. In vitro analysis using murine alveolar cell line MH-S cells primed with LPS showed that BMSCs’ exosomes diminished M1 and promoted M2 markers, which concurred with reduced glycolysis.
Therapeutic potential of BMSC-EXO in LPS-induced ARDS models
2.5.3
BMSC-EXO as a potential therapeutic agent for the treatment of ARDS. This approach is particularly suited to LPS-induced ARDS models, which are commonly used to study sepsis-related lung injury. Experimentally, BMSC-EXO have been illustrated to markedly reduce inflammation and alleviate pathological injury in pulmonary tissues, thereby enhancing lung function. Macrophage polarization is a critical element. M1/M2 amacrophage transition deficiency has devastating effects on the amacrophage. Although multiple biological functions can contribute to this situation, macrophage polarization is the most likely cause, as the LPS-induced switch is designed to increase M1 and reduce M2 inflammatory macrophages. Metabolism is the BMSC, while M1 inflammation has several elaborate metabolic processes.
BMSC-EXO have been demonstrated to carry microRNAs that regulate key cellular processes in alveolar macrophages. A following critical microRNA was miR-384-5p, the levels of which in BMSC-EXO were markedly high. Meanwhile, unlike miR-2909, exosomal miR-384-5p increased, rather than decreased, cell viability. MicroRNA activation occurs not through pyroptosis but by targeting the gene Beclin-1, which plays a critical regulatory role in autophagy. BMSC-EXO are described to alleviate autophagy dysfunction in phagocytes, inhibit apoptosis, and reduce HeLa cell second death. This overall mechanism contributes to the reduction of lung inflammation, the restoration of vascular permeability, and ultimately, to improved survival when BMSC-EXO are administered to LPS-induced ALI rats via intravenous or intratracheal routes (138). In vivo, hBMMSC-exo reduced lung injury scores, decreased inflammation and cell infiltration, and protein leakage in bronchoalveolar lavage fluid, and enhanced survival in septic animals. To our knowledge, this study is the first to show the therapeutic power of BMSC-EXO in modulating macrophage metabolism and inflammation in ARDS. However, the therapeutic potential of MSCs depends primarily on the source of MSCs (139).
Beyond bacterial endotoxin-induced injury, BMSC-EXO have demonstrated efficacy in viral pneumonia models, such as murine cytomegalovirus (CMV)-induced pneumonia. In this context, exosome-treated mice showed a shift in MMSC from an inflammatory M1 to a reparative M2 phenotype, with reduced infiltration of inflammatory cells and improved pulmonary fibrosis. This anti-inflammatory and anti-fibrotic effect of exosomes was due to the downregulation of the NF-κB/NLRP3 inflammasome signaling pathway. We can suggest that the inhibition of macrophage-driven inflammation and fibrotic remodeling may explain how BMSC-EXO attenuate experimental endotoxin-induced ARDS and hold therapeutic potential for patients with viral-induced lung injury (140).
Clinical significance and future directions of macrophage metabolic reprogramming in sepsis-induced ARDS
2.6
Potential of metabolic reprogramming as diagnostic and prognostic biomarkers
2.6.1
Metabolic reprogramming has been identified as a fundamental hallmark in a broad spectrum of diseases. As a result, metabolic reprogramming is a promising biomarker for diagnostic and prognostic applications (141–146). In this regard, metabolic pathways of macrophages, especially glycolysis and OXPHOS, which represent the two polarization states of macrophages, M1 proinflammatory and M2 anti-inflammatory, vary according to the tissue disease gradient and dominate over each other. The dynamic metabolic state of macrophages, encompassing glycolysis, lipid metabolism, and amino acid metabolism, is linked to immune responses, organ failure, and reparative responses in sepsis and COVID-19 (147, 148).
Macrophage polarization is directly linked to metabolic reprogramming. Nowadays, metabolic reprogramming is considered an additional metabolic signature of disease and a marker of responsiveness to existing treatments. In this context, macrophage polarization depends on their glycolytic flux, FAO and amino acid metabolism. At the same time, their polarization has implications for the profile of secreted cytokines. It serves as the basis for the current development of innovative treatments utilizing macrophages in patients with sepsis, cancer, or chronic inflammation (149–152). Moreover, in sepsis: pathogen-induced lung injury (153), metabolic biomarkers of macrophage polarization state are associated with disease severity and therapeutic response to lung-protective mechanical ventilation (153).
The advent of multi-omics technologies, including single-cell RNA sequencing, metabolomics, and proteogenomics, has enabled the identification of highly sensitive and specific biomarkers associated with metabolic reprogramming (154, 155). Significantly, metabolic biomarkers not only indicate the state of disease but also immediately suggest the underlying causes and mechanisms of therapy resistance (156, 157). In sepsis, metabolic shifts in macrophages contribute to immune paralysis and organ dysfunction (158, 159). These insights enable the development of targeted interventions that modulate metabolic pathways to restore immune function and improve clinical outcomes. Metabolism-related molecules and macrophage polarization states represent promising diagnostic and prognostic biomarkers across diverse diseases, including sepsis-induced ARDS. Their dynamic nature allows real-time monitoring of disease progression and therapeutic efficacy.
Targeted therapeutic strategies based on metabolic regulation
2.6.2
Targeting macrophage metabolism has emerged as a promising therapeutic avenue in sepsis-associated ARDS, aiming to restore immune homeostasis by fine-tuning inflammatory responses through metabolic reprogramming. Macrophages, crucial innate immune cells during sepsis and ARDS, shift their metabolism from OXPHOS to glycolysis to meet the increased energy demands of inflammation. Additionally, discovering metabolic advances might sustain the plastic induction of OXPHOS-dependent converting M1.
Compounds such as the natural molecule kaempferol have been effective in treating sepsis-induced ARDS by targeting critical metabolic and inflammatory signaling pathways. As demonstrated by network pharmacology and preclinical-based studies, kaempferol downregulates key pathways, including the HIF-1a, NF-κB, and PI3K-Akt pathways. As a result, proinflammatory cytokines TNF-α, IL-1β, and IL-6 were drastically reduced. In addition to ROS suppression, apoptosis declined in alveolar macrophages. At this point, transcriptional inhibitors of these metabolic pathways that can resolve diseases by restoring immune homeostasis must be activated.
Crucial nodes for intervention are metabolic intermediates and the enzymes that catalyze their reactions. The SphK1/S1PR3 axis promotes glycolysis by upregulating HIF-1α and glycolytic enzymes HK2 and PFKFB3, and it also orchestrates sepsis-induced macrophage polarization into proinflammatory subsets. Pharmacological inhibition of sphingosine kinase 1(SphK1) with PF-543 rebalances macrophage phenotypes by attenuating glycolysis, inhibiting NF-κB activation, and protecting against pathogenic multi-organ damage (29). Closely related to this, enhancement of Spns2/S1P signaling represents a potential approach to neutralize both early hyperinflammation and late-stage immunosuppression by modulating macrophage metabolism and ROS (123). These findings underscore the therapeutic potential of dynamically modulating metabolic regulators to control macrophage function during sepsis.
Itaconate and its derivatives are illustrative of emerging metabolic regulators that contribute as endogenous immunometabolic checkpoints. Itaconate effectively inhibits succinate dehydrogenase, activates the in antioxidant pathway, and modulates glycolytic flux to coordinate the reduction of inflammatory output and oxidative stress in macrophages. Mice models and other preclinical studies have illustrated that itaconate derivatives can reduce cytokine storms and organ injury during sepsis, providing a basis for considering itaconate as a metabolic immunotherapy candidate (160). Moreover, Itaconate derivatives were combined with glycolysis inhibitors in nanoparticle formulations to serve as macrophage-reprogramming interventions, designed to improve the survival of mice with precipitated sepsis. This approach offers a new and creatively engineered modality for metabolic modulation of cells (72).
Therefore, AQPs, and more specifically aquaglyceroporins such as AQP3, AQP7, and AQP9, regulate macrophage metabolism by promoting glycerol transport, which is required for glycolysis and lipid metabolism for immune activation. In addition, AQP targeting also reduces macrophage inflammatory responses and neutrophil unregulated migration in vivo, as evident by reduced inflammation and lung injury in sepsis with AQP9 and AQP5 inhibitors. This renders metabolic substrate transporters attractive hypothetical therapeutic targets for immune cells and inflammation.
Metabolic reprogramming-mediated epigenetic regulation also serves as a therapeutic avenue. Methyltransferase-like 3 (METTL3)-mediated N6-methylation of adenosine in Trim59 mRNA protects against sepsis-induced ARDS by repressing NF-κB signaling and attenuating inflammatory responses. Therefore, targeting the metabolic-epigenetic link can reprogram the function of macrophages and ECs in ARDS (161).
Nanomedicine approaches further enhance metabolic targeting. The “nano-windmill,” a tetrahedral framework nucleic acid-based siRNA delivery system, suppresses macrophage activation by reducing choline uptake, thereby inhibiting glycolysis and inflammatory responses in sepsis models (162). Such precision delivery systems enable targeted modulation of specific metabolic pathways, minimizing off-target effects.
Nanomedicine makes metabolic targeting more sophisticated. A nano-windmill, a tetrahedral framework nucleic acid-based siRNA delivery system, reduces macrophage activation by inhibiting choline uptake, thus preventing glycolysis and inflammation in sepsis models (163). Such targeted delivery technology enables the regulation of metabolic pathways specifically in target cells, thereby preventing off-target side effects. Mitochondrial metabolism plays a quintessential role in macrophage function and is an important intervention target. Promoting mitochondrial biogenesis, proteostasis, and function can endow macrophages with the ability to restore homeostasis and thereby lessen organ injury. Transplantation of exogenous mitochondria has demonstrated an anti-inflammatory and anti-apoptotic effect in ARDS models. Moreover, selenium nanoparticle-modified MSCs facilitate mitochondrial transfer into alveolar epithelial cells and suppress the retinoic acid receptor-related orphan receptor gamma-t (RORγt)/STAT3/Th17 axis in sepsis, thereby reducing inflammatory lung injury (164, 165). Therefore, manipulating lipid metabolism may help restore immunity. Metabolic regulation-based therapeutic modalities targeting macrophages include inhibiting glycolysis and proinflammatory metabolic pathways, enhancing mitochondrial activity, modulating metabolic intermediates and transporters, employing epigenetic measures, and delivering precision nanomedicine. Those strategies allow reprogramming of macrophage polarization, attenuating cytokine storms, decreasing oxidative damage, and restoring immune competence. The heterogeneous and dynamic nature of sepsis and ARDS necessitates a dedicated, personalized approach to multi-targeted metabolism therapies that can achieve a balance between inflammation dampening and immune restoration.
Future research directions and challenges
2.6.3
The intricate crosstalk between metabolic pathways and immune responses in sepsis-induced ARDS represents a critical frontier for future research, with the potential to revolutionize therapeutic approaches. As a result, macrophages, a leading orchestrator of the immune system, rapidly reprogram their metabolism in response to signals, thereby directing their polarization and function. Classical M1-like, proinflammatory macrophages rely on glycolysis for a significant proportion of their fuel, while anti-inflammatory M2-like macrophages prefer OXPHOS and FAO. The imbalances within this structure may exacerbate inflammatory tissue injury or diminish protective mechanisms, increasing the need to unravel these complex networks for medical benefit. M2-like macrophages were transferred to the lungs of mouse models of LPS-induced ALI using exosomes in the BMMSCs (166). The outcomes emphasize immunometabolic treatment, which is effective at the preclinical level and shows responsiveness in signaling pathways; several Mars depend on activation and its transmission, such as HIF-1α, NF-κB, and PI3K-Akt.
Moreover, the metabolic crosstalk is not limited to glycolysis, as the amino acid transporter and utilization also play a role in glutamate transport, which has a protective role in acid-base equilibrium and in reducing oxidative stress in sepsis-induced lung injury, an example of such interaction in cancer that supports a potential approach to use metabolic transporters and enzymes as treatment targets. Simultaneously, the role of aquaporins, especially AQP3 and AQP7, in the transport of glycerol and other small molecules essential for glycolysis and lipid synthesis has also been identified. This involves facilitating AQP3 and AQP7 in regulating macrophage energy demand, regulating inflammatory factor-dependent signaling via TLR4 via the energy-signaling-regulating kinase PI3K, and an excitatory role in macrophage-driven physiological changes. Sepsis treatment by modifying AQPs is efficient, reducing both organ damage and impairments in cellular immune function.
Nevertheless, many obstacles remain. Due to the complex nature of the dynamic sepsis setting, macrophage phenotypes and metabolic states are heterogeneous, making it challenging to search for common therapeutic targets. Moreover, bidirectional interactions between metabolic pathways and immune signaling networks are difficult to decipher without a comprehensive systems biology approach. Nevertheless, machine learning and bioinformatics have identified gene signatures associated with macrophages and mitochondrial biomarkers, both of which can be used to differentiate and individualize patients with sepsis-ARDS (167). However, translating these findings into clinically effective metabolic or cell-based therapies demands rigorous validation and optimization.
Another critical challenge is the temporal dynamics of metabolic reprogramming during sepsis progression. Early metabolic shifts may differ substantially from those in later immunosuppressive phases, necessitating time-sensitive interventions. Impaired alveolar macrophage HSD-1 activity compromises local glucocorticoid activation, exacerbating inflammation and mortality in sepsis-related ARDS, indicating that restoring metabolic enzyme function could enhance endogenous anti-inflammatory responses (75). In addition, retinoic acid deficiency can be considered a syndrome in which autacoids and endogenous metabolic mediators become maladaptive, suggesting that modulation of retinoic acid synthesis could prevent lethal immune dysregulation and cytokine storms in sepsis and ARDS (168).
Future research studies should integrate multi-omics data analysis, complex computational modeling, and confirmatory experimentation to accurately map the metabolic-immune axis. This will enable the production of personalized metabolic interventions, with the potential to deliver metabolic modulators or engineered cytotherapy tuned to metabolism. Additionally, combination therapies that target both metabolic pathways and immune checkpoints could be additive or synergistic.
Conclusion
3
To summarize, the complex metabolic pattern of macrophages in sepsis-associated ARDS is a critical and dynamic course that powerfully modulates and regulates disease development and immune response. From a clinical specialist’s viewpoint, the associations of macrophages, which pass through a “metabolic hurricane” in the early phases and then progress to the second “immune evolution,” illustrate the distinct acclimatizations of macrophage activation and metabolic re-routing. Eventually, this two-phase adaptation not only initiates the inflammatory stage but also determines the balance between an effective immune response and immune system suppression.
The early activation of glycolysis in macrophages is a critical driver of the acute inflammatory response, facilitating rapid energy production and biosynthesis necessary for pathogen clearance. Nevertheless, unchecked high metabolic activity further harms the tissue and exacerbates lung edema. The opposite, the second phase with delayed metabolic activity, underestimates the body’s immune system. In this case, macrophage aggressiveness is significantly reduced, and they are less likely to interfere with reinfections. In most cases, it leads to even serious consequences through the development of secondary insults. Therefore, the metabolic response of cells can be considered a two-component mechanism, implying the opposite regulation of the cell’s metabolic activity.
Emerging evidence on the role of BMMSCs cell-derived exosomes offers promising avenues for therapeutic innovation. By directing macrophage metabolism, exosomes can help maintain immune homeostasis, restrain overactive inflammation, and promote the resolution of lung injury in sepsis-induced ARDS. This development exemplifies a shift towards metabolically focused immunomodulatory interventions rather than traditional, broad-based anti-inflammatory medications.
In a word, future perspectives, the integration of macrophage metabolic profiling into diagnostic frameworks, could enable early identification of patients at risk for immune paralysis or hyperinflammation, allowing for personalized treatment strategies. In addition, the development of pharmacological agents or biologics that selectively modulate macrophage metabolic states offers a more immediate and attainable solution to improve clinical outcomes. However, translating these insights into effective therapies will require rigorous mechanistic studies and well-designed clinical trials to validate safety, efficacy, and the optimal timing of interventions. Ultimately, balancing various research perspectives, from molecular metabolism to immunology and regenerative medicine, will be instrumental in advancing our understanding and management of sepsis-associated ARDS. The interplay between metabolic pathways and immune function in macrophages is complex and highly context-dependent, necessitating a multidisciplinary approach to dissect. Future research focused on understanding the precise signaling networks and metabolic checkpoints that govern macrophage polarization and function within the septic lung environment may provide the necessary insights to develop therapeutic targets.
In conclusion, macrophage metabolic reprogramming plays a vital role in shaping the inflammatory or immunological microenvironment of ARDS. The ability to leverage this breakthrough and develop metabolism-centered clinical or molecular therapy strategies is a forward-looking approach with huge potential to revolutionize patient care. Future studies must bridge the existing discrepancy and incorporate new technologies in order to fully translate the health implications of macrophage metabolic therapy in this crippling disorder.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Wang Z Wang Z . The role of macrophages polarization in sepsis-induced acute lung injury. Front Immunol. (2023) 14:1209438. doi: 10.3389/fimmu.2023.1209438, PMID: 37691951 PMC 10483837 · doi ↗ · pubmed ↗
- 2Liu SY Ruan H Li SS . HIF-1α: a bridge connecting sepsis and acute respiratory distress syndrome. Eur J Med Res. (2025) 30:827. doi: 10.1186/s 40001-025-03107-z, PMID: 40887582 PMC 12399009 · doi ↗ · pubmed ↗
- 3Wang QL Yang L Liu ZL Peng Y Gao M Deng LT . Sirtuin 6 regulates macrophage polarization to alleviate sepsis-induced acute respiratory distress syndrome via dual mechanisms dependent on and independent of autophagy. Cytotherapy. (2022) 24:149–60. doi: 10.1016/j.jcyt.2021.09.001, PMID: 34920961 · doi ↗ · pubmed ↗
- 4Qiu YJ Zhan F Cheng HP Shao M Li XH Bao XW . Targeting Glutamate transport: A breakthrough in mitigating sepsis lung injury. Free Radic Biol Med. (2025) 235:190–9. doi: 10.1016/j.freeradbiomed.2025.04.043, PMID: 40294854 · doi ↗ · pubmed ↗
- 5Li Q Zheng H Chen B . Identification of macrophage-related genes in sepsis-induced ARDS using bioinformatics and machine learning. Sci Rep. (2023) 13:9876. doi: 10.1038/s 41598-023-37162-5, PMID: 37336980 PMC 10279743 · doi ↗ · pubmed ↗
- 6Zhang S Liu Y Zhang XL Sun Y Lu ZH . ANKRD 22 aggravates sepsis-induced ARDS and promotes pulmonary M 1 macrophage polarization. J Transl Autoimmun. (2024) 8:100228. doi: 10.1016/j.jtauto.2023.100228, PMID: 38225946 PMC 10788270 · doi ↗ · pubmed ↗
- 7Zhu Z Liu L . Exploring the potential role of the cholinergic anti-inflammatory pathway from the perspective of sepsis pathophysiology. J Intensive Care Med. (2025) 40:571–80. doi: 10.1177/08850666251334342, PMID: 40223326 · doi ↗ · pubmed ↗
- 8Wang H Xu P Yin K Wang S . The role of m 6A modification during macrophage metabolic reprogramming in human diseases and animal models. Front Immunol. (2025) 16:1521196. doi: 10.3389/fimmu.2025.1521196, PMID: 40066451 PMC 11891544 · doi ↗ · pubmed ↗