Netherton Syndrome: A Systematic Review of the Challenges of Diagnosis and Treatment
Maximus S Reese, Ryan Nguyen, Noor Chughtai, Philip J Haynos, Salma Alkhatib, Shantanu Amin, Ethan Speer, Jared Nichols

TL;DR
Netherton syndrome is a rare genetic skin disorder with challenges in diagnosis and treatment, requiring more research for effective therapies.
Contribution
This paper systematically reviews 30 case reports to highlight diagnostic and therapeutic challenges in Netherton syndrome.
Findings
Biologic therapies improved symptoms and infection rates but did not lead to complete remission.
Genetic testing is the most reliable diagnostic method but is not always accessible.
Treatment outcomes vary, emphasizing the need for individualized care and larger studies.
Abstract
Netherton syndrome is an autosomal recessive genodermatosis caused by biallelic mutations in the SPINK5 gene, a gene that codes for lymphoepithelial Kazal-type-related inhibitor 1 (LEKT1) protein. This disorder is characterized by erythroderma, ichthyosis, hair shaft abnormalities, and immune dysregulation. While there is increasing research and recognition of Netherton syndrome, much remains unknown, with limited knowledge of the history, manifestations, and responses to therapies. The purpose of this study was to review case reports of Netherton’s to draw conclusions from patient demographics, symptoms, treatment, and outcomes. A systematic review of 30 case reports and clinical studies on Netherton syndrome was conducted, including only patient-level clinical data of studies in English. Data were extracted via full-text review, organized into comparative tables, and analyzed for…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
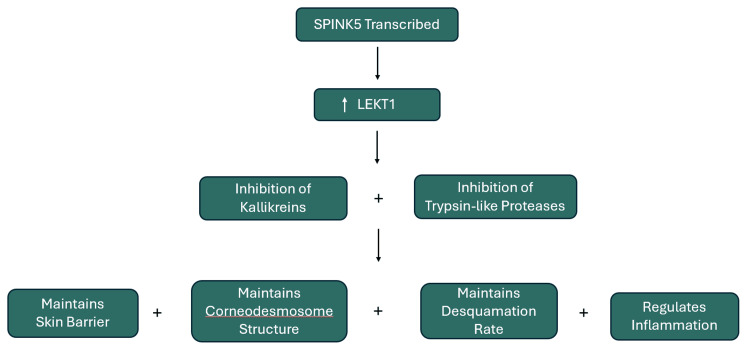 Figure 1
Figure 1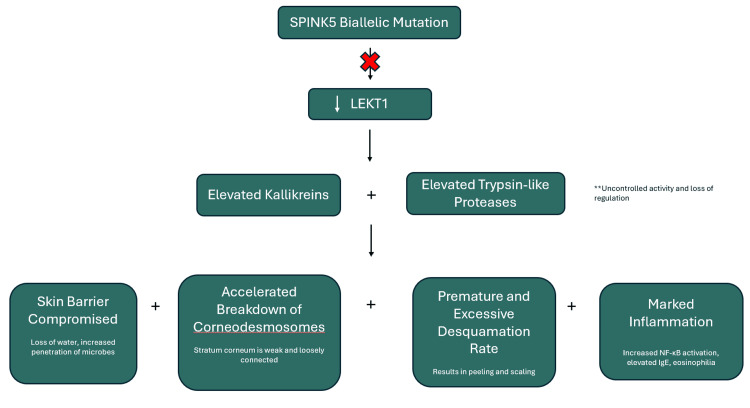 Figure 2
Figure 2| Clinical symptom | Number of cases presenting |
| Erythroderma/reddish skin/generalized erythema | 34 |
| Hair abnormalities (general: thin, fragile, sparse, brittle, lusterless, short)- “Bamboo Hair”-trichorrhexis invaginata | 27* |
| Scaling/desquamation | 16 |
| Recurrent infections | 15 |
| Pruritus | 18 |
| Atopic manifestations (allergies, asthma) | 17 |
| Ichthyosis linearis circumflexa (ILC) | 10 |
| Diagnostic category | Common methods used |
| Genetic testing | SPINK5 sequencing, WES, qRT-PCR, Sanger sequencing |
| Hair analysis | Light/electron microscopy, trichoscopy |
| Skin biopsy | Psoriasiform hyperplasia, parakeratosis, spongiosis |
| Immunohistochemistry | LEKTI, IL-36, cytokine markers |
| Lab tests | IgE ↑, eosinophils ↑, vitamin D ↓, WBC ↑ |
| Imaging | Chest X-ray, bone age, echo, brain MRI |
| Immune/allergy testing | Flow cytometry, IgE panels |
| Misdiagnosis cases | Atopic dermatitis, ichthyosis, PRP |
| Diagnostic finding | Number of cases presenting |
| Elevated IgE | 28 |
| Genetic testing confirming SPINK5 mutation | 26 |
| Immunohistochemistry staining for LEKTI | 6 |
| Hyperplasia | 6 |
| Hyperkeratosis | 10 |
| Spongiosis | 13 |
| Treatment type | Number of cases using treatment | Cases with reduced symptoms | Cases with failed treatment | Adverse effects |
| Immunosuppressants (topical corticosteroids) | 22 | 8 | 14 | Immune system suppression; increased infection risk |
| Antihistamines | 9 | 5 | 4 | Sedation |
| Antibiotics (systemic or topical) | 8 | 5 | 3 | GI upset; vulnerable to certain infections |
| Dupilumab | 8 | 5 | 3 | No adverse effects noted in adult or infant |
| Omalizumab | 1 | 1 | 0 | No adverse effects noted |
| Infliximab | 1 | 1 | 0 | No adverse effects noted |
| Pembrolizumab | 1 | 1 | 0 | In previous studies, pembrolizumab showed various adverse effects involving multiple organ systems. In this report, no adverse events were noted |
| Secukinumab* | 6 | 4 | 2 | A potential adverse effect from treatment was noted in one of the cases; however, it was later found negligible due to the same findings in the absence of treatment. The other cases of secukinumab treatment involved no adverse effects |
| Nutritional support/ supplements/allergen-free diet | 8 | 5 | 3 | No adverse effects noted |
| Acitretin (oral retinoid) | 5 | 2 | 3 | No adverse effects noted |
| Abrocitinib | 1 | 1 | 0 | No adverse effects noted |
| Tofacitinib | 1 | 0 | 0 | No adverse effects noted |
| Upadacitinib | 1 | 1 | 0 | No adverse effects noted |
| Other specific treatments (acyclovir, clotrimazole) | 3 | 3 | 0 | No adverse effects noted |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSkin and Cellular Biology Research · Wnt/β-catenin signaling in development and cancer · Marine Invertebrate Physiology and Ecology
Introduction and background
Netherton syndrome (NS) is a rare, autosomal recessive genodermatosis disorder caused by biallelic mutations in the SPINK5 gene. This gene encodes the protein lymphoepithelial Kazal-type-related inhibitor 1 (LEKT1), a serine peptidase inhibitor that controls the activity of certain serine peptidase enzymes responsible for the breakdown of other proteins in the outer layer of the skin (stratum corneum) [1]. LEKT1 also plays a role in normal hair follicle development, the development of lymphocytes, and the control of peptidases that trigger immune system function. Mutations in SPINK5 result in decreased LEKT1 allowing for increased breakdown of proteins in the epidermis resulting in excessive skin shedding, abnormal hair growth, and immune dysfunction [1,2]. A summary of the SPINK5 gene's role can be seen in Figure 1.
SPINK5 gene pathway. LEKT1: lymphoepithelial Kazal-type-related inhibitor 1.
Netherton's is classically characterized by erythroderma (diffuse, red skin at birth or early infancy), ichthyosis (scaly skin), hair shaft abnormalities (trichorrhexis invaginata, “bamboo hair”), and immune dysregulation (elevated IgE, eosinophilia, recurrent infections). NS can become severe due to life-threatening electrolyte imbalances, dehydration, or because of the recurrent infections, which exhibit that this disease can have a significant impact on quality of life [3-5].
Being a rare disease, much remains unknown about why NS manifests, what therapies best work for it, and how to best distinguish it from other severe dermatological disorders. Pathophysiology is poorly understood, and its clinical phenotype is variable. Many symptoms overlap with other severe dermatological disorders, which complicates diagnosis. While there are no current standardized therapies, accurate identification can rule out inappropriate treatment options and help patients receive care that may be beneficial sooner. Molecular genetic testing for the SPINK5 mutation is known to be key in confirming NS, but resources are variable, and this test cannot always be ordered. Presently, much of the treatment for NS is supportive, but emerging targeted therapies have potential in individualized and successful care. Further study and research is needed to better understand disease mechanisms and develop evidence-based treatment strategies [6]. A depiction of the SPINK5 gene mutation pathway is seen in Figure 2.
SPINK5 gene mutation pathway. LEKT1: lymphoepithelial Kazal-type-related inhibitor 1.
Review
Methods
A systematic review of existing case reports and clinical studies involving patients with Netherton syndrome was performed with a focus on patient-level data. A total of 34 patients from 30 studies were included, with articles being retrieved from the PubMed database. The search term "Netherton syndrome" was used with filters limiting results to published case reports within the past 10 years. This search retrieved 52 articles, of which 30 were ultimately selected based on their relevance and variability in treatment approaches to allow for cross-analysis. Additional reports of Netherton syndrome were identified but excluded due to incomplete clinical data. Relevant case reports may exist that were not included; therefore, the findings of this review should be considered for the sample collected.
Inclusion criteria for this review required studies with patient-level clinical data on subjects, including the patient’s medical history, presenting symptoms, previous treatments, diagnostic methods, treatments administered, and patient outcomes. Exclusion criteria included non-English reports without translation and papers lacking sufficient clinical detail of patient history, presentation, or diagnosis. To extract data for this report, full-text reviews were performed for each article considered. Data were organized into comparative tables and categories based on presentation, diagnostic techniques, lab findings, and treatment. Findings for the review were synthesized by identifying common patterns across reports, diagnostic challenges highlighted, exhibited treatment effectiveness, and important physician insights noted in managing this disease. All included studies were case reports; however, no formal risk-of-bias or quality assessment was performed. Potential bias and limitations of this review are considered in the interpretation of the findings.
Results
Thirty-four patients were identified across 30 case reports, including those from studies reporting multiple affected individuals. Patient ages ranged from approximately one month to 62 years, the age gap reflecting when patients were looking for definitive treatment. Gender distribution was as follows: 23 males (67.65%) and 11 females (32.35%). Two adult Bahraini siblings (58 year old female and 62 year old male), a 27 month old Omani male, and a seven year old Algerian female were reported in the data to be born to consanguineous parents -- these cases highlighted the autosomal recessive inheritance pattern of Netherton Syndrome (11.76% of cases presented from first degree consanguineous parents) [7-9]. Patients were globally distributed, representing the Middle East, Asia, Europe, and North America. Tables 1-4 summarize the analysis of reviewed cases, including presentations, lab findings, treatments, and key findings (see Appendix, Table 5).
Certain findings were more frequently observed, including erythroderma, trichorrhexis invaginata, skin scaling, and atopic features [10-12]. Trichorrhexis invaginata was seen in 27 cases (79.41%). Atopic manifestations, such as asthma and allergies, were observed in 17 patients (50%). Desquamation was described in 16 cases (47.06%), alopecia in one case (2.94%), and ichthyosis linearis circumflexa (ILC) in 10 patients (29.14%). Erosions, hyperpigmentation, onychogryphosis, ectropion, vesicles, body odor, and vulvovaginal involvement were inconsistent findings whose manifestation was influenced by coexisting diseases [13,14].
Diagnostic methods included: genetic testing in 26 patients (76.47%), immunostaining of LEKT1 for six cases (17.65%), microscopy (hair or skin) in 12 cases (35.29%), and trichoscopy in four cases (11.76%) [15-18]. Access to advanced diagnostics varied from case to case. Initial misdiagnosis of NS occurred in four cases (11.76%).
Dupilumab was administered to eight patients (23.53%), secukinumab in six cases (17.65%), and pembrolizumab was used in one case (2.94%). Intravenous immunoglobulin (IVIG) therapy in nine patients (26.47%), omalizumab in two cases (5.88%), and JAK inhibitors (abrocitinib, tofacitinib, upadacitinib) in three cases (8.82%) with reported clinical improvement (reduced erythema) [19-30]. Supportive care involved topical corticosteroids, emollients, antihistamines, tacrolimus, and antibiotics (64.70%). Nutrition support for diet control and supplements was used for eight cases (23.52%). Acitretin was orally used for five cases (14.70%). Other specific treatments used in three cases (8.82%), such as acyclovir and clotrimazole, were seen in use due to associated diseases. Standard treatment is not yet established for this disease. Biological therapies, dupilumab and secukinumab, as well as IVIG, showed an association with symptom improvement across multiple cases of moderate to severe NS. However, none of the treatments explored achieved durable remission, and long-term efficacy data are limited. Use of emollients, corticosteroids, and antibiotics is important in symptom management, but does not provide treatment.
Clinical improvement was defined as reduced erythema, reduced itchiness, reduced scaling and dry skin, improved hair structure, and treatment of infections. Improvements were observed across various medication courses, but without a standardized treatment for NS, each case report consisted of a different regimen. Fourteen cases showed there were no signs of improvement (41.18%); eight cases (23.53%) showed relapse into the initial presentation, while six cases (17.64%) showed no reaction to treatments. There were cases that switched treatment due to failure to resolve symptoms, while others did not note treatment going forward. No patients achieved total curative remission, and no deaths were identified across the cases reviewed [31].
Discussion
Genetic confirmation of biallelic variations of SPINK5 gene mutations remains the most reliable diagnostic tool; however, limited access restricts use [2]. In cases without genetic confirmation, the importance of recognizing distinguishing hallmark features of NS through detailed physical exams proved to be critical in order to avoid misdiagnosis with other dermatopathological diseases.
Alternative diagnostic methods included LEKTI and IL-36 immunostaining, which did aid in supporting diagnosis, but both were insufficient in providing a diagnosis individually [16]. LEKT1 technique demonstrated the protein’s deficiency in affected NS individuals, and IL-36 staining indicated that Th17-driven inflammation was occurring, but these techniques do not replace genetic confirmation, as they are more unreliable in definitive diagnosis. With other frequent findings, a similar situation is faced. Hair shaft abnormalities, particularly trichorrhexis invaginata, are a very valuable diagnostic finding, as the majority of cases showed this finding, but the absence of trichorrhexis invaginata does not exclude NS (atypical presentations exist). Scaling, desquamation, pruritus, and recurrent infections present due to the compromised skin barrier and immune dysfunction that characterize the disease, but they too are not reliable in making a diagnosis alone. Due to misdiagnosis of NS with other atopic dermatopathies, these findings work in congruence with one another in support of NS diagnosis; none of these findings alone is sufficient for diagnosis.
Clinical presentation of NS can vary reflecting the heterogeneity of dermatologic involvement within this disease; however, it was identified that erythroderma and eosinophilia presented in every patient reviewed. Elevated IgE is non-specific to NS, but does explain other symptoms that can occur, such as asthma, allergies, erythroderma, hair shaft defects, and atopic diathesis. Less common findings included ichthyosis linearis circumflexa (ILC) and skin manifestations, such as erosions, ectropion, and hyperpigmentation. Variability of presentation highlights the difficulty of diagnosis of NS [25].
Severe complications consisted of symptoms that progressed from dermatologic involvement. Underlying barrier dysfunction and immune dysregulation can lead to recurrent infections, electrolyte imbalances, and failure to thrive. Hypernatremia hospitalized an infant due to electrolyte imbalances, and such severe progression of NS could lead to a fatal outcome if not managed properly and early [19-21]. Recurrent infections along with a failure to thrive were reported, and while non-specific to NS, they can lead to mortality as well. One report described a squamous cell carcinoma (SCC) case, and while evidence is currently insufficient to state if there is an elevated malignancy risk, this occurrence shows a potential malignancy and morbidity risk. Therefore, oncologic vigilance is reasonable in long-term dermatologic treatment of NS, but the reviewed case reports do not provide sufficient data to establish protocols [30].
Current treatment of NS remains focused on symptomatic relief; however, the use of biologic agents described in this review shows promise, though data on long-term efficacy are limited. These findings emphasize the need for larger studies with standardized outcome reporting in order to establish treatment guidelines. Variability in NS presentation and treatment response highlights that early diagnosis and counseling is especially important in avoiding NS progressing to more severe forms that can be life-threatening. Individualized therapy and genetic counseling still seem to be the course of action for this disease.
Limitations of this study include a small sample size, variations in diagnostic techniques, heterogeneity of outcomes for patients, and failure to follow up/follow-up duration. Potential biases include detection, selection, and reporting. Fatal cases may be under-reported, while surviving cases may be disproportionately studied in higher detail. These constraints restrict the generalizability of findings, but do highlight the need for further studies and standardized reporting for improvement in NS treatment.
Conclusions
Netherton's syndrome was shown to consistently present with erythroderma, ichthyosis, hair shaft abnormalities, and immune dysregulation to varying degrees. Complications of Netherton's, such as electrolyte imbalances, dehydration, or recurrent infections, will increase the severity of this disease, emphasizing the need for early identification and diagnosis. Molecular testing remains the most effective diagnostic modality, with a detailed physical exam remaining the mainstay diagnostic tool in low-resource areas. Symptomatic management with topical emollients, corticosteroids, and antibiotics has been successful with the currently proposed treatment therapies, which include biologics (dupilumab and secukinumab) and IVIG. However, these treatment options have varied in their lasting success.
Due to its rarity, NS remains shrouded in relative obscurity, and as such, early accurate diagnosis and consequently treatment remains a challenge. This systematic review used a 34-patient sample size to identify common NS presentations, successful diagnostic modalities, and successful therapies. The review proposes a pathway-dependent individualized therapy in union with genetic counseling as potentially the most reliable route to successful treatment. The review encourages future research endeavors with a larger sample size and a focus on lasting treatments.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Netherton syndrome with a novel likely pathogenic variant c. 420del (p. Ser 141Profs Ter 5) in SPINK 5 gene: a case report Case Rep Dermatol Kovacheva K Kamburova Z Vasilev P Yordanova I 4716202410.1159/000536083 PMC 1089080838406644 · doi ↗ · pubmed ↗
- 2Netherton syndrome caused by heterozygous frameshift mutation combined with homozygous c. 1258 A> G polymorphism in SPINK 5 gene Genes Moltrasio C Romagnuolo M Riva D 10801420233723944010.3390/genes 14051080 PMC 10218509 · doi ↗ · pubmed ↗
- 3A novel mutation in SPINK 5 gene underlies a case of atypical Netherton syndrome Front Genet Wang Y Song H Yu L Wu N Zheng X Liang B Wang P 9432641320223615998910.3389/fgene.2022.943264 PMC 9500337 · doi ↗ · pubmed ↗
- 4Severe hypernatremia as presentation of Netherton syndrome Glob Med Genet Di Nora A Consentino MC Messina G Timpanaro T Smilari P Pavone P 335338102023 https://journal.hep.com.cn/gmg/EN/10.1055/s-0043-17769833802519510.1055/s-0043-1776983 PMC 10665120 · doi ↗ · pubmed ↗
- 5Netherton syndrome associated to Candida parapsilosis otomycosis BMJ Case Rep Merad Y Derrar H Ouldsaid K Benallel K 014202110.1136/bcr-2021-243260 PMC 829272634285026 · doi ↗ · pubmed ↗
- 6Protease and protease-activated receptor-2 signaling in the pathogenesis of atopic dermatitis Yonsei Med J Lee SE Jeong SK Lee SH 8088225120102087904510.3349/ymj.2010.51.6.808PMC 2995962 · doi ↗ · pubmed ↗
- 7A novel pathogenic variant in the corneodesmosin gene causing generalized inflammatory peeling skin syndrome with marked eosinophilia and trichorrhexis invaginata Pediatr Dermatol Gordon H Yap P Hsiao KC Watson M Purvis D 2682723920223517875210.1111/pde.14939 PMC 9305742 · doi ↗ · pubmed ↗
- 8Two incidental sibling diagnoses of Netherton syndrome in separate visits: a case report Cureus Al Moosawi S Alkhanaizi S Albaharna M Khamdan F 016202410.7759/cureus.56439 PMC 1102476238638763 · doi ↗ · pubmed ↗