ERGA-BGE genome of Dendarus foraminosus: an IUCN Least Concern darkling beetle endemic to Crete (Greece)
Giannis Bolanakis, Danae Karakasi, Apostolos Trichas, Astrid Böhne, Rita Monteiro, Rosa Fernández, Nuria Escudero, Eleftherios Bitzilekis, Manos Stratakis, Petros Lymberakis, Nikolaos Poulakakis, Manon Angel, Manon Angel, Jean-Marc Barbance, Julie Batisse, Odette Beluche

TL;DR
This paper presents the genome of Dendarus foraminosus, a darkling beetle endemic to Crete, to support evolutionary and population studies.
Contribution
The study provides a high-quality chromosome-level reference genome for Dendarus foraminosus.
Findings
The genome assembly includes 11 chromosomal pseudomolecules with a total size of 0.59 Gb.
The assembly has high contig and scaffold N50 values of 24.4 Mb and 51.9 Mb, respectively.
Abstract
Dendarus foraminosus Mulsant and Rey, 1855 is a darkling beetle in the family Tenebrionidae and one of the many Dendarus species endemic to the island of Crete. Dendarus foraminosus is a commonly found species and is widespread in the lowland and montane phrygana and maquis of central Crete. The species is classified as Least Concern (LC) by the IUCN Red List. The reference genome of Dendarus foraminosus will enable phylogenetic, population, and evolutionary research regarding this endemic species and its close relatives. A total of 11 contiguous chromosomal pseudomolecules (sex chromosomes included) were assembled from the genome sequence. This chromosome-level assembly encompasses 0.59 Gb, composed of 430 contigs and 415 scaffolds, with contig and scaffold N50 values of 24.4 Mb and 51.9 Mb, respectively.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1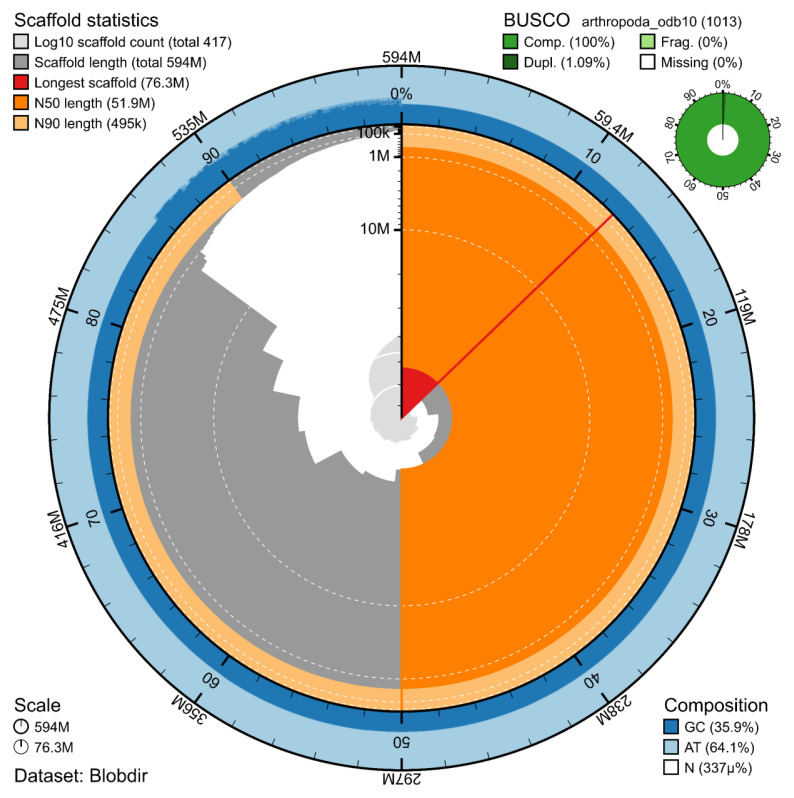 Figure 2
Figure 2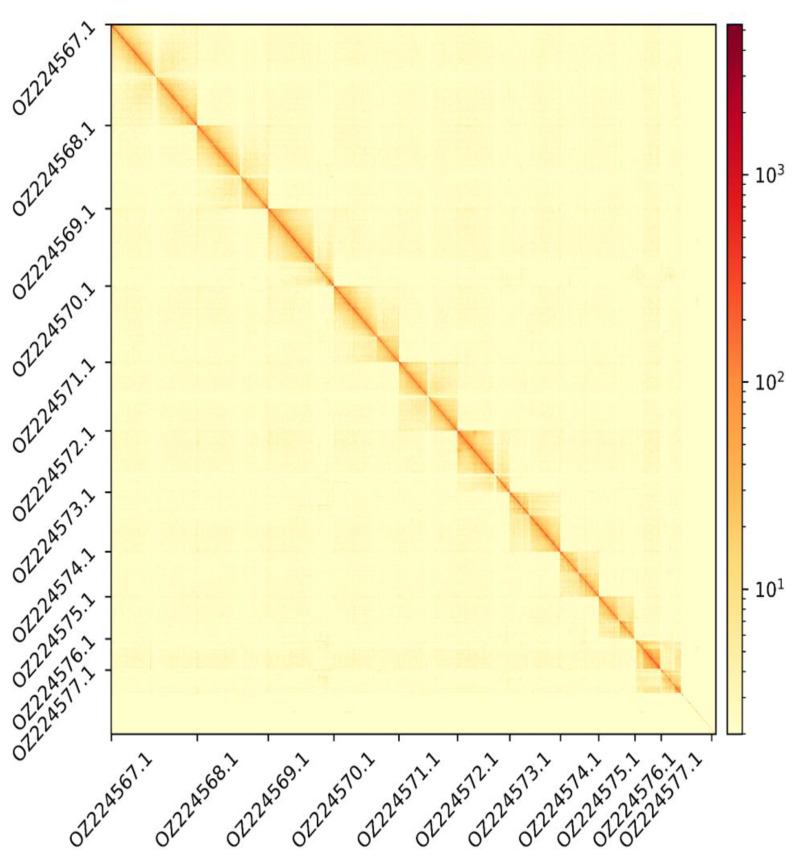 Figure 3
Figure 3| No.
| No.
| Mean gene
| No. single-exon
| Mean exons
| |
|---|---|---|---|---|---|
|
| 15,992 | 23,634 | 10,478 | 554 | 5.3 |
|
| 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
|
| 22 | 22 | 116 | 22 | 1.0 |
|
| 1,864 | 1,978 | 4,892 | 3 | 2.2 |
|
| 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
|
| 71 | 17 | 136 | 71 | 1.0 |
|
| 77 | 77 | 692 | 77 | 1.0 |
|
| 1 | 1 | 128 | 1 | 1.0 |
|
| 300 | 300 | 74 | 300 | 1.0 |
|
| 3 | 3 | 298 | 3 | 1.0 |
| Complete | Single copy | Duplicated | Fragmented | Missing | |
|---|---|---|---|---|---|
|
| 981 (96.9%) | 969 (95.7%) | 12 (1.2%) | 8 (0.8%) | 24 (2.3%) |
|
| 4,465 (93.7%) | 4,175 (87.6%) | 290 (6.1%) | - | 298 (6.2%) |
| Consistent | Inconsistent | Contaminants | Unknown | ||
|
| 12,671 (79.2%) | 339 (2.1%) | 0.0 (0.0%) | 2,982 (18.6%) | |
- —UK Research and Innovation (UKRI)
- —Horizon Europe Framework Programme
- —Agence Nationale de Recherche
- —Agence Nationale de Recherche
- —Swiss State Secretariat for Education, Research and Innovation (SERI)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsForest Insect Ecology and Management · Forest Ecology and Biodiversity Studies · Coleoptera Taxonomy and Distribution
Introduction
Dendarus foraminosus is a darkling beetle in the family Tenebrionidae. The species is endemic to the island of Crete ( Trichas, 2008) and is mostly found in the central parts of the island, from lowland phrygana and maquis to montane shrublands. In the west- and the east-side of the island, the species is replaced by its congeneric species D. opacus and D. puncticollis respectively, while in the higher altitudes of Psiloritis and Dikti mountains by D. politus. All these species together form the Dendarus Cretan lineage ( Trichas et al., 2020). According to the IUCN Red List (Red List Necca 2025, unpublished assessment), the species is not threatened by extinction, as it occurs in great abundance in many localities and various habitats in Crete.
The species larvae are saprophytophagous, participating in the decomposition of the rotting plant matter.
The reference genome of D. foraminosus will help in understanding the phylogenetic relationships of the species of the genus Dendarus in Crete, the species delimitation of the Cretan lineage, the recovery of its population structure, and the species adaptation in the Cretan environment in the face of climate change.
The generation of this reference resource was coordinated by the European Reference Genome Atlas (ERGA) initiative’s Biodiversity Genomics Europe (BGE) project, supporting ERGA’s aims of promoting transnational cooperation to promote advances in the application of genomics technologies to protect and restore biodiversity ( Mazzoni et al., 2023).
Materials & Methods
ERGA's sequencing strategy includes Oxford Nanopore Technology (ONT) and/or Pacific Biosciences (PacBio) for long-read sequencing, along with Hi-C sequencing for chromosomal architecture, Illumina Paired-End (PE) for polishing (i.e. recommended for ONT-only assemblies), and RNA sequencing for transcriptome profiling, to facilitate genome assembly and annotation.
Sample and sampling information
On April 9th, 2022, 11 male adults of Dendarus foraminosus were sampled by Giannis Bolanakis ( Figure 1). The species was identified by the expert entomologist Dr. Apostolos Trichas. The specimens were hand-picked in Mount Kedros, Rethymno, Crete (Greece). Sampling was performed under the Presidential Degree 67/81 of the Greek Government. The specimens were euthanized in liquid nitrogen and were preserved at -80°C until DNA extraction.
A darkling beetle ( Dendarus foraminosus) from a different specimen from the one sampled for this study.The specimen was photographed on the island of Crete. Photo credit: Dr. Apostolos Trichas.
Vouchering information
Physical reference material for the here sequenced specimen has been deposited in the Arthropods Collections of the Natural History Museum of Crete of the University of Crete (NHMC) https://www.nhmc.uoc.gr/en/departments/arthropods/ under voucher ID NHMC.85.2.26267.
Frozen reference tissue material of abdomen, head and pronotum of D. foraminosus have been deposited in the Genomics and Genetic Resources Division of the NHMC https://www.nhmc.uoc.gr/ under voucher ID NHMC.85.2.26267.
Genetic information
The estimated genome size, based on ancestral taxa, is 0.41 Gb. This is a diploid genome with a haploid number of 10 chromosomes (2n = 20). All information for this species was retrieved from Genomes on a Tree ( Challis et al., 2023).
DNA/RNA processing
DNA was extracted from a whole individual (80 mg) using a conventional CTAB extraction followed by a commercial purification using Qiagen Genomic tips (QIAGEN, MD, USA). A detailed protocol is available on protocols.io ( https://www.protocols.io/view/hmw-dna-extraction-for-long-read-sequencing-using-bp2l694yzlqe/v1).
DNA fragment size selection was performed using Short Read Eliminator (PacBio, CA, USA). Quantification was performed using a Qubit dsDNA HS Assay kit (Thermo Fisher Scientific) and integrity was assessed in a FemtoPulse system (Agilent). DNA was stored at 4 °C until usage. RNA was extracted using an RNeasy Plus Universal Kit (Qiagen) following manufacturer instructions. RNA was extracted from the whole individual (80 mg) and extracted RNA was then treated with 6U of TURBO DNase (2 U/μl) (Thermo Fisher Scientific). Quantification was performed using a Qubit RNA HS Assay and integrity was assessed in a Bioanalyzer system (Agilent). RNA was stored at -80 °C.
Library preparation and sequencing
Long-read DNA libraries were prepared with the SMRTbell prep kit 3.0 following manufacturers' instructions and sequenced on a Revio system (PacBio). Hi-C libraries were generated from the whole individual using the Dovetail Omni-C Kit (following the Insects & marine invertebrates’ protocol v1.2) and sequenced on a NovaSeq 6000 instrument (Illumina) with 2x150 bp read length. Poly(A) RNA-Seq libraries were constructed using the Illumina Stranded mRNA Prep, Ligation Prep kit (Illumina) and sequenced on an Illumina NovaSeq 6000 instrument.
Genome assembly methods
The genome of Dendarus foraminosus was assembled using the Genoscope GALOP pipeline ( https://workflowhub.eu/workflows/1200). Briefly, raw PacBio HiFi reads were assembled using Hifiasm v0.19.5-r593. Retained haplotigs were removed using purge_dups v1.2.5 with default parameters and the proposed cutoffs. The purged assembly was scaffolded using YaHS v1.2 and assembled scaffolds were then curated through manual inspection using PretextView v0.2.5 to remove false joins and incorporate sequences not automatically scaffolded into their respective locations within the chromosomal pseudomolecules. Two scaffolds with low coverage, and homology with Tenebrio molitor (X, Y) chromosomes, were renamed as X and Y respectively. The Telomeric repeat pattern found is TCGGG. Chromosome-scale scaffolds confirmed by Hi-C data were named in order of size. The mitochondrial genome was assembled as two circular forms using Oatk v1.0 and included in the released assembly. Summary analysis of the released assembly was performed using the ERGA-BGE Genome Report ASM Galaxy workflow ( https://doi.org/10.48546/workflowhub.workflow.1104.1).
Genome annotation methods
A gene set was generated using the Ensembl Gene Annotation system ( Aken et al., 2016), primarily by aligning publicly available short-read RNA-seq data from BioSample SAMEA112751342 to the genome. Gaps in the annotation were filled via protein-to-genome alignments of a select set of clade-specific proteins from UniProt ( UniProt Consortium, 2019), which had experimental evidence at the protein or transcript level. At each locus, data were aggregated and consolidated, prioritising models derived from RNA-seq data, resulting in a final set of gene models and associated non-redundant transcript sets. To distinguish true isoforms from fragments, the likelihood of each open reading frame (ORF) was evaluated against known metazoan proteins. Low-quality transcript models, such as those showing evidence of fragmented ORFs, were removed. In cases where RNA-seq data were fragmented or absent, homology data were prioritised, favouring longer transcripts with strong intron support from short-read data. The resulting gene models were classified into two categories: protein-coding, and long non-coding. Models that did not overlap protein-coding genes and were constructed from transcriptomic data were considered potential lncRNAs. Potential lncRNAs were further filtered to remove single-exon loci due to their unreliability. Putative miRNAs were predicted by performing a BLAST search of miRBase ( Kozomara et al., 2019) against the genome, followed by RNAfold analysis ( Gruber et al., 2008). Other small non-coding loci were identified by scanning the genome with Rfam ( Kalvari et al., 2018) and passing the results through Infernal ( Nawrocki & Eddy, 2013).
Results
Genome assembly
The genome assembly has a total length of 593,991,149 bp in 415 scaffolds including two circular forms of the mitogenome ( Figure 2 and Figure 3), with a GC content of 35.9%. It features a contig N50 of 24,404,000 bp (L50 = 9) and a scaffold N50 of 51,873,200 bp (L50 = 5). There are 15 gaps, totalling 2.0 kb in cumulative size. The single-copy gene content analysis using the Arthropoda database with BUSCO ( Manni et al., 2021) resulted in 100% completeness (98.9% single and 1.1% duplicated). 71.3% of reads k-mers were present in the assembly and the assembly has a base accuracy Quality Value (QV) of 63.4 calculated by Merqury ( Rhie et al., 2020).
Snail plot summary of assembly statistics.The main plot is divided into 1,000 size-ordered bins around the circumference, with each bin representing 0.1% of the 593,991,149 bp assembly including the mitochondrial genome. The distribution of sequence lengths is shown in dark grey, with the plot radius scaled to the longest sequence present in the assembly (76.3 Mb, shown in red). Orange and pale-orange arcs show the scaffold N50 and N90 sequence lengths (51,873,200 bp and 494,690 bp), respectively. The pale grey spiral shows the cumulative sequence count on a log-scale, with white scale lines showing successive orders of magnitude. The blue and pale-blue area around the outside of the plot shows the distribution of GC, AT, and N percentages in the same bins as the inner plot. A summary of complete, fragmented, duplicated, and missing BUSCO genes found in the assembled genome from the Arthropoda database (odb10) is shown on the top right.
Hi-C contact map showing spatial interactions between regions of the genome.The diagonal corresponds to intra-chromosomal contacts, depicting chromosome boundaries. The frequency of contacts is shown on a logarithmic heatmap scale. Hi-C matrix bins were merged into a 100 kb bin size for plotting. On both axes the GenBank names of the 11th largest autosomes are shown.
Genome annotation
The genome annotation consists of 15,992 protein-coding genes with associated 23,634 transcripts, in addition to 2,335 non-coding genes ( Table 1). Using the longest isoform per transcript, the single-copy gene content analysis using the Arthropoda odb10 database with BUSCO resulted in 96.9% completeness. Using the OMAmer Metazoa-v2.0.0.h5 database for OMArk ( Nevers et al., 2025) resulted in 93.7% completeness and 79.2% consistency ( Table 2).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Aken BL Ayling S Barrell D : The Ensembl gene annotation system. Database (Oxford). 2016;2016: baw 093. 10.1093/database/baw 093 27337980 PMC 4919035 · doi ↗ · pubmed ↗
- 2Challis R Kumar S Sotero-Caio C : Genomes on a Tree (Goa T): a versatile, scalable search engine for genomic and sequencing project metadata across the eukaryotic Tree of Life [version 1; peer review: 2 approved]. Wellcome Open Res. 2023;8:24. 10.12688/wellcomeopenres.18658.1 36864925 PMC 9971660 · doi ↗ · pubmed ↗
- 3Gruber AR Lorenz R Bernhart SH : The Vienna RNA websuite. Nucleic Acids Res. 2008;36(Web Server issue):W 70–W 74. 10.1093/nar/gkn 188 18424795 PMC 2447809 · doi ↗ · pubmed ↗
- 4Kalvari I Nawrocki EP Argasinska J : Non-coding RNA analysis using the Rfam database. Curr Protoc Bioinformatics. 2018;62(1):e 51. 10.1002/cpbi.51 29927072 PMC 6754622 · doi ↗ · pubmed ↗
- 5Kozomara A Birgaoanu M Griffiths-Jones S : mi R Base: from micro RNA sequences to function. Nucleic Acids Res. 2019;47(D 1):D 155–D 162. 10.1093/nar/gky 1141 30423142 PMC 6323917 · doi ↗ · pubmed ↗
- 6Manni M Berkeley MR Seppey M : BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol. 2021;38(10):4647–4654. 10.1093/molbev/msab 199 34320186 PMC 8476166 · doi ↗ · pubmed ↗
- 7Mazzoni CJ Ciofi C Waterhouse RM : Biodiversity: an atlas of European reference genomes. Nature. 2023;619(7679):252. 10.1038/d 41586-023-02229-w 37433931 · doi ↗ · pubmed ↗
- 8Nawrocki EP Eddy SR : Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29(22):2933–2935. 10.1093/bioinformatics/btt 509 24008419 PMC 3810854 · doi ↗ · pubmed ↗