HIV-associated sensory neuropathy: current progress and future challenges
Isaac J. Gamez, Ajay Pal, Connor Haines, Subo Yuan

TL;DR
This paper reviews the current understanding and challenges in treating HIV-associated sensory neuropathy, highlighting the need for better treatments and research.
Contribution
The paper provides a comprehensive review of recent developments and challenges in managing HIV-associated sensory neuropathy.
Findings
No FDA-approved treatments exist for HIV-associated sensory neuropathy.
Adjusting antiretroviral therapy is a primary management strategy.
Research is needed to understand the mechanisms of HIV-SN for better treatment.
Abstract
Currently, no US Food and Drug Administration-approved treatments exist to manage HIV-associated sensory neuropathy (HIV-SN) and management is largely confined to adjusting antiretroviral therapy (ART) doses and medications. Thus, this urgent health crisis requires strong research commitment to identify a cure or palliative treatment. This review explores the current state-of-the-art related to HIV-SN. It first explores recent developments in the understanding of HIV-SN, emphasizing the importance of developments in the HIV-SN mouse model and non-myelination intra epidermal never fiber denervation. Next, the neurotoxic side effects of ART are summarized. Finally, we explore the interactions and synergy between HIV-SN and ART in the pathogenesis of peripheral neuropathy. While the overall mortality related to HIV has decreased significantly in recent decades, further elucidation of the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
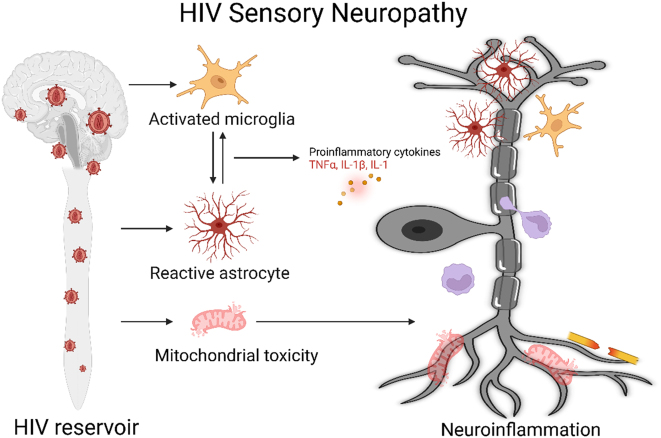 Figure 1
Figure 1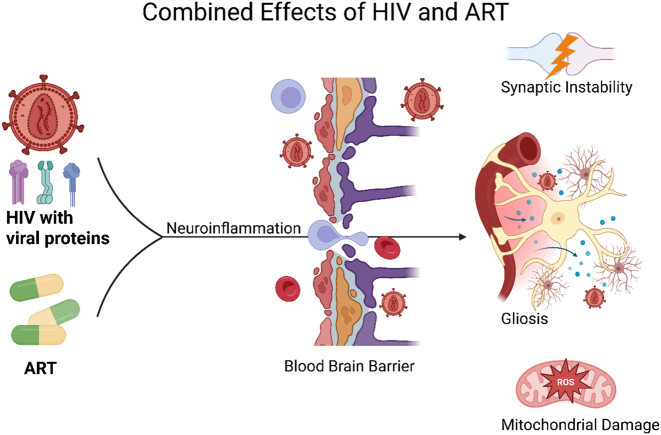 Figure 2
Figure 2| Agent | Mechanism | Clinical impact | Potential therapeutic intervention |
|---|---|---|---|
| Tat | Activates NMDAR → ↑Ca2+ influx → mitochondrial dysfunction and neuron apoptosis. | Neuronal hyperexcitability, peripheral sensitization, and neuropathic pain. | NMDA receptor antagonists and mitochondrial protectants. |
| Vpr | Increases neuronal Ca2+→ mitochondrial damage; triggers TNF-α–mediated inflammation. | Neuronal hyperexcitability, and inflammation. | Calcium channel modulators and anti-inflammatory agents. |
| gp120* | Binds receptors/coreceptors CD4, CXCR4, CCR5 in glia cell →activation of microglial & astrocyte cells→ neuroinflammation. | Allodynia in gp120 rodent pain model, chronic HIV-PAIN+/SN and other neurological disorders. | CCR1 antagonists, microglial inhibitors, NMDA blockers, and Wnt5a antagonists. |
| ART (NRTIs) | Mitochondrial toxicity and dysfunction → upregulation of Wnt5a → astrocyte activation and neuroinflammation → axonal degeneration. | Neurotoxicity, and induced peripheral neuropathy. | Replace with less neurotoxic agents, mitochondrial protectants, and Wnt5a antagonists. |
| CNS Viral reservoir | Latent infection in macrophages and microglia → continued secretion of viral proteins despite ART. | Chronic neuronal injury, and chronic neuroinflammation. | Latency-reversing agents, BBB-penetrant ARTs, and use of nanoparticle vehicles. |
- —University of Texas Medical Branch, Center for Interdisciplinary Research in Women's Health (CIRWH) and Sealy Center on Aging (SCOA)
- —National Institute of Neurological Disorders and Stroke
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHIV Research and Treatment · HIV-related health complications and treatments · Facial Nerve Paralysis Treatment and Research
Introduction
By the end of 2023, the Joint United Nations Programme on HIV/AIDS and the World Health Organization reported that an estimated 39.9 million people worldwide live with HIV [1]. Global access to antiretroviral therapy (ART) has turned HIV infection from a deadly disease into a controllable, chronic disease and closed the gap in lifespan between people living with HIV infection (PLWH) and the general population [2]. Unfortunately, the prevalence of HIV associated pain (HIV-PAIN) does not dramatically decrease when ART has effectively suppressed the HIV viral viremia [3]. The presence of under-addressed HIV-PAIN, including HIV associated sensory neuropathy (HIV-SN), is demoralizing, dulls quality of life and, importantly, disrupts ART adherence [4].
The difficulties of chronic HIV infection are not only related to immune deficiency but also to neurological sequelae such as peripheral neuropathy, cerebrovascular disease, and neurocognitive impairment [5], [6], [7]. In the progression of HIV infection, there is an increased risk of HIV-SN due to residual viral mediated neuroinflammation [8], [9], [10], [11], [12]. HIV-SN ranges in clinical presentation from numbness and tingling all the way to debilitating neuropathic pain that can interfere with activities of daily living, resulting in social consequences such as unemployment [13], 14]. Furthermore, the utilization of ART to treat HIV counterproductively induces sensory neuropathy through neurotoxic side effects [15], [16], [17]. Indeed, HIV-SN has an estimated 30–60 % prevalence in HIV infected patients, varying by countries and region [18]. For example, some estimates of HIV-SN are as low as 8.6 % [16] in the United States and as high as 42 % [19] in Australia. High prevalence of disease is reported in African countries with rates reaching as high as 36 % [20]. The cause of discrepancy between countries is multifactorial as delays in diagnosis, access to newer antiviral medications, and opportunistic infection rates differ by region [20]. In the post ART era, prevalence increases with advanced age, female gender, and advanced stage of HIV infection, with ART treatment itself aggravating existing neuropathy [20], [21], [22], [23], [24].
Abnormal nociception
Physiological somatosensation begins in the skin with specialized receptors that detect touch, temperature, proprioception, and chemicals [25], 26]. Mechanoreceptors and free nerve endings transmit nociceptive signals through dorsal root ganglion (DRG) neurons to the spinal dorsal horn (SDH). Secondary afferent neurons enter the central nervous system (CNS) and ascend to higher centers like the thalamus and cerebellum [26]. Dysfunction along this pathway can lead to neuropathic pain, defined as pain from lesions or disease of the somatosensory system. Peripheral neuropathy involves distal-to-proximal “dying-back” axonal degeneration, triggered by chemical insults, trauma, or disrupted axonal transport, often involving calcium influx and axon-destructive processes [27]. Incomplete nerve repair can cause peripheral sensitization, presenting as spontaneous pain, allodynia, and hyperalgesia, with reduced intraepidermal nerve fiber density correlating with pain severity [27]. Persistent peripheral input may lead to central sensitization, where CNS neurons become hyper-responsive, manifesting as wind-up, secondary hyperalgesia, and chronic pain. Central neuropathic pain is associated with conditions such as stroke, Parkinson’s disease, spinal cord injury, and multiple sclerosis [28].
HIV-associated sensory neuropathy (HIV-SN)
HIV cannot directly infect neurons due to lack of CD4 receptor[29]. However, many mechanisms exist by which HIV influences neuronal health. The typical life cycle of HIV begins with virion entry into CD4+ cells, which includes T cells and monocytes. The binding of gp120 to CD4+ receptors facilitate gp120 further binding to coreceptor CCR5 and CXCR4. This process mediates HIV viral entry into the cell. The initial infection phase involves integration of viral cDNA into host DNA through integrase activity [30]. Infected cells harbor integrated viral DNA and may be transcriptionally silent forming the latent viral reservoir. The post integration phase includes viral protein production, release of an immature virion and subsequent maturation, as well as secretion of viral proteins even in the absence of virus [30]. Immune cells make an ideal sanctuary for HIV, as they are widespread amongst tissues, have prolonged lifespans, and have diverse interactions with other cell types. Monocytes and macrophages exert influence on peripheral nerves and cause neuroinflammation as described below. Additionally, HIV infected monocytes (CD14+ and CD16+) transmigrate across the blood-brain barrier (BBB), resulting in CNS invasion and neuronal compromise. This activity represents a major issue in targeting HIV infection, as the CNS acts as a hidden reservoir. Even in full suppression of HIV peripherally, transcription continues to occur in the CNS, and viral reservoirs are found within the brain. The viral reservoir is protected not only from peripherally circulating pharmaceutical agents, including ART, but also from immunorecognition and elimination due to the brain’s immune privilege. This persistent, low-level expression of viral proteins contributes to ongoing neuronal dysfunction and damage, particularly in the context of chronic neuroinflammation. We will first discuss the direct mechanisms by which HIV proteins, including Tat, Viral protein R (Vpr) and gp120, cause neuronal damage. Then, we will discuss the major role of neuroinflammation as mediated by glial cell types to induce synaptic degeneration and subsequent neuropathy.
As indicated, HIV-SN occurs even in individuals with long-term controlled viremia. Most of the neuronal toxicity has been associated with the secretion of viral proteins from viral reservoirs into neighboring uninfected cells such as neurons [12], [31], [32], [33]. In agreement, exposure to the viral protein Tat for more than 48 h induced DRG apoptosis in rat models [31]. Tat can activate N-methyl-D-aspartate receptors (NMDAR) that subsequently signal for neuron apoptosis through Ca^2+^ influx [34]. Disruption of Ca^2+^ homeostasis has toxic effects on mitochondria which results in chronic energy deficits leading to abnormal electrical discharges and neuronal degeneration [35], 36]. This association with neuronal hyperexcitability is an important link to the peripheral sensitization, which contributes to neuropathy. HIV Vpr similarly induces mitotoxic effects on human DRG. Vpr induces a transient increase of Ca^2+^ in neurons leading to hyperexcitability and ultimately neuronal toxicity [9]. Additionally, Vpr is associated with neuroinflammation, as indicated by higher levels of TNF-α [9], 11]. Although the neurotoxic effects of Tat and Vpr are known, the overall contribution in HIV-SN remains uncertain. Much of the evidence implicating Tat and Vpr derives from high-concentration exposure studies that may not reflect physiologic levels in chronically infected patients [9], 31]. Furthermore, postmortem analyses have not consistently demonstrated higher levels of Tat and Vpr expression in the SDH of HIV-PAIN^+^ patients compared with HIV-PAIN^−37^. This discrepancy suggests that the relevance of Tat and Vpr in vivo may depend upon other factors. Overall, the current body of evidence favors gp120 as a primary driver of HIV-SN, whereas the contributions of Tat and Vpr require further investigation for clarification.
Gp120 has been implicated as a major inducer of HIV-SN [32], 37]. Gp120 expression is significantly higher in the SDH spinal cord, a pain processing center where peripheral nociceptive signals integrate into the CNS. Specifically, gp120 levels in HIV pain-positive patients were 10 times higher in the SDH than those in pain-negative patients [37]. Additionally, markers of synaptic degeneration are elevated in HIV patients with pain [12], 37], and this result is reproducible in gp120 pain mouse models via intrathecal injections (i.t.) of gp120 [37].
Epidermal protein gene product 9.5 (PGP9.5) innervation and HIV-SN
Healthy epidermis of skin innervated with nociceptors (sensory neuronal terminals) functions to detect external stimulation such as temperature, chemical, touch, vibration and mechanical stimulation. These nociceptors can be immunopositively labeled by protein gene product 9.5 (PGP9.5^+^) in the dermis and epidermis [37], 38]. PGP 9.5^+^ was first recognized as a brain-specific protein decades ago, richly expressed in different neurons in vertebrates and forming an estimated 5–10 % of the cytoplasmic protein [39]. Within the epidermis area, only unmyelinated nociceptors (C-fiber) detect nociceptive signals and transduce nociception [40]. Pan et al. and Karlsson et al. reported that degeneration of intraepidermal nerve fiber density (IENF) occurred in both distant extremities, more obvious in distant leg, and that degeneration preceded the elevation of thermal thresholds (decreased sensitivity to heat) in neuropathy patients [41], 42]. PGP9.5^+^ is commonly used to label peripheral nerve fibers and the degeneration of the PGP9.5^+^ cutaneous nociceptor is a representative pathological biomarker of various sensory neuropathies, including HIV-SN [43], 44]. HIV-SN patients and HIV-PAIN rodent models usually associate persisting, chronic pain with progressing degeneration or even denervation of skin native PGP9.5^+^ nociceptors. According to Zhou and colleagues, IENF at the distant leg site correlates with neuropathy severity as gauged by total Neuropathy Score, a reflection of the severity of distal symmetric polyneuropathy [45]. Shi et al. reported that sural nerve autopsy showed 5/5 patients with intense ART treatment had pathological diagnosed neuropathy [46]. Additionally, Ebenezer et al. reported that the sural nerve biopsy of HIV-SN patients showed nerve fiber loss predominantly in distal terminal, including both the epidermis (IENF) and dermis, with the IENF loss being prior to loss of larger caliber and deeper dermal axons [47]. Shikuma et al. found that IENF decreases in the distal leg of HIV negative patients (30.1^#^/mm) compared to IENF in HIV positive patients with NRTI treatment (21.1^#^/mm) with further decline accompanying treatment with NRTIs [48]. These works suggest the IENF in distant limbs are the initial site of degeneration by HIV infection with ART treatment and are generally recognized as a key pathological biomarker of HIV-SN. A critical challenge is that ART cannot directly prevent IENF reduction. Previously, Mangus et al. investigated the pathogenesis of HIV-SN using simian immunodeficiency virus (SIV)-infected Asian macaques. At 84 days post viral infection, the length of PGP9.5+ nerve fiber were diminished, as measured through footpad skin biopsies [49]. This work indicates that both HIV and SIV can induce similar SN. Human samples prove the reality of HIV-SN, whereas SIV-SN was used as a close mimic of human HIV-SN. However, both human HIV and macaque SN provides limited opportunities for mechanistic analysis. For deep HIV-SN pathogenesis investigation, rodents, especially the mouse HIV-SN model, are more convenient for detailed mechanism research. Yuan et al. replicated HIV-PAIN and SN in mice by i.t. gp120 and observed that PGP9.5^+^ IENF in hind paw glabrous skin exhibited temporal degeneration post gp120 i.t, thus the mice were named as mHIV-SN mouse model. At three weeks, the innervation of plantar eccrine glands showed simultaneously the same patten of degeneration as hind paw skin, suggesting that mice developed autonomic neuropathy as well. Corresponding with the development of allodynia and PGP9.5^+^ pathologies, the envelope glycoprotein of HIV, gp120, plays an essential role in targeting SDH and sensory neurons both in humans and mice. The resulting degeneration of PGP9.5^+^ nociceptors is associated with both mouse and human HIV-PAIN and HIV-SN [37].
HIV-SN mouse model
Yuan et al. developed mHIV-SN by i.t. of HIV-1 gp120Bal and gp120IIIB, two coreceptor tropism of gp120. I.t. injection makes gp120 easily diffuse to lumbar enlargement, an epidural area where L4, 5, 6 DRGs and spinal nerves are located and create a scenario that mimics the gp120 increase in the SDH, DRG, and lumbar spinal nerve of hHIV-SN [50]. This is the first mouse model parallel of gp120-induced nociceptive pathologies in mice and patients [37], 51]. This i.t. gp120 mHIV-SN phenocopies the behavioral, neurological, glial, synaptic, and molecular pathologies observed in human HIV-SN (hHIV-SN). Pain is a major comorbidity of hHIV-SN and has been clearly observed in mHIV-SN. Of particular importance, the mHIV-SN model develops robust ‘dying-back’ SN, manifested as denervation of PGP9.5^+^ C-fiber nerve endings in the epidermis and autonomic neuropathy, evidenced by neuroinflammation and denervation of the sweat gland. This is clinically aligned with observations that hHIV-SN is often accompanied by autonomic neuropathy [37]. Additionally, the SDH tissue from mHIV-SN and hHIV-SN show extensive pathological similarities, suggesting the relevance of mHIV-SN to hHIV-SN [37]. The strength of the mHIV-SN model to date is that pain pathologies have been confirmed to systematically phenocopy pathologies of hHIV-SN and HIV-PAIN patients. Acharjee et al. investigated the mechanisms of a cytotoxic HIV-1 accessory protein, Vpr, on human DRG neurons. Vpr caused DRG neuronal damage, rising neuronal calcium level and cytokine perturbation, suggesting its contribution to HIV-SN and HIV-PAIN [9], [52], [53], [54]. Keswani and colleagues, using didanosine on gp120 transgenic mice that expressed gp120 under a Glial Fibrillary Acidic Protein promoter, found that such mice developed distal degeneration of unmyelinated sensory axons, and C-fiber loss as seen in patients with HIV-SN [52]. Wallace et al. revealed that rats with perineural HIV-gp120 treated with zalcitabine (i.p. 50 mg/kg) showed degeneration of C fibers in the epidermis and allodynia [53]. Together, these studies suggest that both HIV-SN and NRTIs induce SN caused C-denervation, which mainly manifests as HIV-PAIN [53].
Pathogenic role of HIV viral protein
The gp120 glycoprotein on the HIV-1 envelope mediates virus entry by binding to a CD4 receptor. Despite effective ART, neurological abnormalities persist in the post-ART era [24]. Our HIV postmortem data indicate that the development of HIV-SN associated pain was intimately linked with extraordinary high levels of gp120 protein in SDH in hHIV-SN patients (3/5) with pain even on intensified ART, compared to relatively lower levels of gp120 in hHIV-SN patients (2/5) with pain but not on ART. Additionally, levels of both the Tat and Vpr proteins were much lower than gp120 in all hHIV-SN patients with pain [37], indicating that ART can effectively the decrease viral load and improve CD4 cell counts, but cannot stop the persistent gp120 expression in SDH. This observation shows the key etiological role of gp120 (rather than viral load per se) in hHIV-SN, as plasma viral loads and CD4 cell counts are not closely associated with hHIV-SN [37], 55]. Moreover, gp120 transgenic mice provide in vivo evidence that gp120 is responsible for HIV associated nervous system impairment [56], including neuropathic pain beginning at 4 months [12]. Overall, gp120 is the critical viral protein for HIV to induce HIV-SN, a critical nociceptive mechanism for HIV-PAIN both in mice and humans.
Soluble gp120 similarly exerts effects on neurons through NMDAR binding [12], 33]. Gp120 directly binds to NMDAR, triggering a signaling cascade that upregulates fractalkine and its receptor CX3CR1, leading to activation of microglia [12]. It has been further elicited that gp120 induced fractalkine upregulation is mediated by the Wnt3a/beta-catenin signaling pathway [12]. Activated microglia then contribute to synaptic degeneration, as shown by synaptic protein loss in vitro and in vivo [12]. Activated microglial cells secrete inflammatory products such as IL-1β, TNF-α, and brain-derived neurotropic factors. While microglial activation plays a significant role in the early phases of gp120-induced neuropathic pain, it is less involved in late phases [57], 58]. This is evidenced by similar levels of microglial markers, such as ionized Ca^2+^ binding adaptor molecule 1 and CD11b, in the SDH of HIV negative and HIV positive groups when evaluating chronic pain [57]. The transient nature of microglial activation is observed in rodent models post-nerve injury, with microglial proliferation beginning to decline after 7 days [59]. This result suggests that microglia do most of their damage in the acute phase of inflammation, but not chronically. This is indeed evident with microglial ablation only partially inhibiting the early phase of gp120 induced hyperalgesia without impacting the late stage of pain development in mouse models [58]. (Table 1, Figures 1 and 2).
HIV reservoirs induce HIV associated sensory neuropathy through mechanisms of neuroinflammation. HIV viral proteins such as gp120, Tat and Vpr contribute to neuroinflammation via activation of astrocytes and microglia. Neurons, astrocytes, and microglia have crosstalk via expression of inflammatory cytokines. HIV viral proteins are also toxic to mitochondria of neurons. Illustration created in https://BioRender.com.
Combined effects of HIV and antiretroviral therapy (ART). The effects of HIV, including viral proteins, and ART combine to generate neuroinflammation through the Wnt5a pathway. Additionally, neuroinflammation disrupts the integrity of blood brain barrier (BBB) and permits the entry of immune cells, and HIV into the tightly regulated central nervous system (CNS). Viral protein and ART drug-induced neuroinflammation cause synaptic instability, gliosis, and damage to mitochondria. Illustration created in https://BioRender.com.
The non-canonical pathway utilizing the Wnt family member 5a (Wnt5a) has been shown to be a major signal molecule in gp120 HIV-SN [60]. In this pathway, gp120 upregulates Wnt5a in neurons [58]. The release of Wnt5a activates astrocytes utilizing the receptor tyrosine kinase-like orphan receptor 2, among other receptors [58]. Now that astrogliosis is achieved, pathways including c-Jun N-terminal kinase and calcium/calmodulin-dependent protein kinase II are used to stimulate neuroinflammatory molecules [8], 37], 60]. Wnt5a expression specifically induces IL-1β, IL-1, and TNF-α, leading to neuroinflammation [8], 10]. This overexpression of inflammatory markers recruits circulating monocytes to the sites of damage and induces apoptosis, causing destruction to nearby neurons [61]. Gp120 additionally induces neural circuit polarization, whereby increased inputs to excitatory neurons and reduced excitation in inhibitory neurons cause central sensitization. This central sensitization is facilitated through astrogliosis and IL-1β and is associated with chronic pain [58].
Other cell types, including DRG resident macrophages, oligodendrocytes, and Schwann cells, have also been implicated in gp120 induced neuroinflammation, but the literature is limited on their roles in the pathogenesis of HIV-SN [12], [62], [63], [64].
Antiretroviral therapy (ART) induced neuropathy
Nucleoside reverse transcriptase inhibitors (NRTIs) compete with natural deoxynucleotides for incorporation into a growing viral DNA chain. However, NRTIs lack a 3′-hydroxyl group on the deoxyribose moiety. This difference results in incorporating an NRTI, and the next incoming deoxynucleotide cannot form the following 5′, 3′ phosphodiester bond needed to extend the DNA chain. The result is a chain termination in DNA synthesis [65]. The usage of dNTP is a common feature of all DNA polymerases, and thus NRTIs expected to block human as well as the viral DNA polymerases and lead to toxicity [66], [67], [68]. ART, the only FDA-approved treatment for HIV infection, is composed of a cocktail of medicines targeting different stages of the viral cell cycle. A typical HIV regimen of ART generally include the “backbone” of two NRTIs and one additional class of ART [65]. Other ART classes include an integrase inhibitor, a non-nucleoside reverse transcriptase inhibitor (NNRTI) or a protease inhibitor. ART does not cure HIV, but dramatically increase life expectancy of HIV patients and is crucial for lowering the risk of transmission [2]. Rapid initiation of ART is associated with better health outcomes in HIV patients [69]. While ART therapy is a mainstay in treatment of HIV, usage of certain ART is still limited due to ART related toxicity.
ART dramatically extends the life span, decreases comorbidities and improves the quality of life of PLWH; however, life-long administration of certain ART causes neurological complications, including SN. NRTIs are especially relevant in this regard, because their neurotoxicity is well observed both in patients [70], replicated NRTI administration induced neuroinflammation in the mouse brain [71], and in the spinal cord [72]. According to the US Centers for Disease Control and Prevention, in a cohort of 2,515 PLWH on ART treatment, 329 (13.1 %) were diagnosed with HIV-SN [73]. However, the incidence may be sharply underreported. In our study, 5/5 postmortem HIV PAIN patients were pathologically diagnosed with HIV-SN by sural nerve autopsy [46]. The relative contributions of unresolved inflammation, ongoing HIV replication, and ART-related neurotoxicity may all contribute, and clinical evidence cannot definitively identify either viral infection or ART toxicity as the primary driver of HIV-SN.
Viral proteins have been shown to preferentially damage large Aβ-fiber myelinated primary afferent axons that convey touch and proprioceptive information, whereas NRTIs preferentially damage small myelinated Aδ fibers and nonmyelinated C fibers that convey temperature and pain information [43]. Peripheral neuropathy is a painful and debilitating complication associated with NRTI therapy. The dideoxy-NRTIs zalcitabine and didanosine, as well as the thymidine-analog NRTI stavudine may hinder adherence to the life-saving treatment [74], 75]. Early NRTIs, including zalcitabine, now are discontinued for clinical use. NRTIs are well-documented to induce mitochondrial dysfunction and oxidative stress. Currently HIV treatment regimens, such as Biktarvy (bictegravir/emtricitabine/tenofovir alafenamide) contain an integrase strand transfer inhibitor and two backbone medicine of NRTIs, tenofovir, emtricitabine. While current ART regimens have lower levels of toxicity than older NRTI drugs, they are still reported to induce neuropathy in clinic. Tenofovir causes peripheral neuropathy in patients [73] as well as in wild type and gp120 transgenic mice, with alterations in inflammatory signaling and mitochondrial activity in neurons [76]. Tenofovir and emtricitabine also showed neurotoxicity, with the concentration related to the drug’s reported CSF concentration level [77]. Emtricitabine is part of several recommended first-line ART, one-pill a day regiments. Emtricitabine combines with other classes of ART drugs such as Biktarvy (bictegravir/emtricitabine/tenofovir alafenamide), Atripla (efavirenz/emtricitabine/tenofovir disoproxil fumarate), Delstrigo (doravirine/lamivudine/tenofovir disoproxil fumarate) [78]. Emtricitabine is found to induce peripheral neuropathy, especially when used in combination with tenofovir. However, Margolis assessed that both emtricitabine and tenofovir are less common to cause mitochondrial toxicity compared with older NRTIs [70]. The transition to newer generation ART agents with more favorable safety profiles has also extended to HIV pre-exposure prophylaxis (PrEP) regimens such as Truvada (emtricitabine/tenofovir disoproxil fumarate), Descovy (emtricitabine/tenofovir alafenamide), and Apretude (cabotegravir). Although limited evidence suggests that PrEP usage may be associated with modest increases in immune activation [79], comprehensive safety reviews report no clinically meaningful risk of neuropathy in PrEP users [80]. This is further supported by the rare prevalence of neuropathic symptoms in large PrEP cohorts [81]. However, our preliminary data of Biktarvy (emtricitabine, tenofovir alafenamide, bictegravir) showed induction of chronic pain and neuropathy in mice (unpublished data). Currently, HIV-SN is still with significant prevalence in an era where HIV viremia is dramatically suppressed. We cannot exclude the possibility that the newer generation of ART is free from neurotoxicity and ART associated neuropathy, as the NRTIs, such as emtricitabine and tenofovir alafenamide, are still the major components of most ART polypills. Therefore, the underlying unknown pathogenesis of ART in addition to mitochondrial toxicity needs to be explored further. ART neurotoxicity studies are limited and have a long way to go.
Mitochondrial dysfunction and neuroinflammation are major mechanisms by which ART promotes neuropathy. DNA polymerase α, δ, and ε are insensitive to inhibition by dideoxynucleotides, but both DNA polymerases β and γ can be inhibited in vitro by these compounds [68], 74], 75], [82], [83], [84], [85], [86], [87]. As DNA polymerase γ is the only DNA polymerase involved in mitochondrial DNA (mtDNA) replication, the impact of NRTIs on this enzyme can adversely affect mitochondrial replication and function, leading to dysfunction [66], 82], [88], [89], [90]]. Studies in cell lines [91], 92], animal models [93], [94], [95], and in humans [96], [97], [98] have shown morphologic, quantitative, and functional changes of mitochondria following NRTI exposure. The mitochondrial dysfunction mechanism has been investigated for many years, with the “DNA poly-gamma hypothesis” the most prominent. This hypothesis posits that NRTIs adversely affect mitochondrial replication through binding to enzyme DNA polymerase γ and terminating DNA chain formation [17], 99]. Blockage of this protein leads to mtDNA depletion, disruption of ATP production, and increased production of free reactive oxygen species [100]. While the details of this cascade have not been fully characterized, the report of a novel, functional mtDNA POLG mutation being associated with NRTI-associated lactic acidosis [101] lends weight to the POLG inhibition hypothesis [102]. This is clinically correlated with the significant depletion of mtDNA and abnormal levels of electron transport chain proteins found in the DRG of HIV pain positive patients as opposed to HIV patients without neuropathic pain [103]. Furthermore, mitochondria represent a major metabolic hub with important connections to neuroinflammation (see review by Wang et al. [104]). Disruption in mitochondrial metabolism leads to metabolites that strongly stimulate inflammation and activate macrophages [104]. Changes to the mitochondrial membrane potential permit reactive oxygen species to accumulate leading to induction of IL-1β and mitochondria autophagy [104]. Theoretically, mitochondrial damage affects all organs and components over time (except adult red blood cells), but the inherent heterogeneity in drug absorption and mitochondrial activity dynamics make tissues with the highest energy demand most susceptible. As the nervous system is one of the most energy consuming, it is thus severely affected [105], [106], [107]. DRGs are particularly vulnerable due to their limited regenerative capacity and high metabolic demand. The defect in replication machinery ultimately impacts the oxidative phosphorylation system leading to complications that may include neurological manifestations such as peripheral neuropathy, encephalopathy, dementia, seizures, and stroke [96].
Besides mitochondrial dysfunction, other mechanisms also contribute to ART induced chronic pain and SN. Yuan et al. reported that NRTI (specifically, zalcitabine, 3′-azido-3′-deoxythymidine [AZT], lamivudine, and stavudine) administration for two weeks induced mice allodynia, with zalcitabine upregulating Wnt5a expression (SDH). Additionally observed was activation of astrocyte and microglia in the spinal cord dorsal horn of 15.5-month-old mice, which approximates 50 years in humans [108]. The mouse study aligns with that of Lichtenstein et al., who found that, in PLWH treated with NRTI regiment, being >40 years was a significant risk factor for HIV-SN. This work indicates that NRTI elicited Wnt5a mediated neuroinflammation, and inhibition of Wnt5a with Box5 blocked zalcitabine induced up-regulation of TNF-alpha, IL-1beta, and IL-6. Thus, spinal cord neuroinflammation critically contributes to zalcitabine induced HIV-SN [30]. Using a creative drosophila larvae model to investigate NRTI-induced SN, Bush et al. modeled AZT induced allodynia and thermal hyperalgesia in drosophila larvae fed with AZT. They found that AZT exposure increases the dynamics and instability of sensory neurons measured through terminal dendrite sprouting, growing, and retracting on live imaging. Restoring dendritic stability through Par-1 knockdown suppresses the fragmentation-like phenotype induced by AZT in flies [109]. This work built an excellent model in understanding the mechanisms of ART induced SN and provides insight on the mechanism of NRTI disrupting stability of sensory neruons [109]. Synapse stability is another factor by which ART can influence neuronal health even before the death of a neuron. As seen in other neurodegenerative diseases such as dementia, the lack of synaptic stability of dendrites indicates neuronal dysfunction [108]. Indeed, improving synapse stability in the context of NRTI induced neuropathy led to a suppression of nociception hypersensitivity in a drosophila model [109]. Synapse dysfunction is measured by synapse turnover rates, post-synaptic plasticity, and impaired circuit function [108]. AZT, an NRTI, causes synaptic instability and fragmentation of sensory dendrites [110]. Synaptic instability and dynamic dendrites are other mechanisms by which NRTI causes neurotoxicity and subsequent neuropathy.
Combined toxicity of HIV and ART
We now explore the synergistic effects of HIV and ART in initiating neuropathy. How ART induces neuropathy is difficult to ascertain, as it may unmask subclinical HIV neuropathy or indeed add directly to neurotoxicity. In any case, ART administration has been shown to be a risk factor for developing neuropathy and those with mild neuropathy may have progressive disease in a dose-dependent manner [16], 111]. In patients with mild peripheral neuropathy, usage of NRTIs, including zalcitabine, didanosine, stavudine, and lamivudine, exacerbated symptoms [112]. The need for therapeutic strategies that minimize these adverse effects while effectively managing HIV infection is critical. So far, the mainstay in symptomatic treatment of neuropathic pain due to ART is dose adjustment or switching medication regimens. While planning a medication regiment to avoid the most neurotoxic medications is feasible in the United States, patients in poorer countries must still utilize older ARTs due to cost.
HIV-SN, a major pathology of HIV-PAIN, can be aggravated by NRTIs which are the backbone of ART. There are two critical algogens, gp120 and NRTIs which are tightly intermingled in HIV-PAIN and are impossible to untangle. Yuan et al. identified gp120 as a major algogenic protein in HIV-PAIN by comparing the HIV-PAIN mortem SDH and mouse spinal cord [37]. Shi Y et al. found that gp120 up-regulated Wnt5a expression in the mortem spinal cord of HIV-PAIN patient and the mouse spinal cord [54], 113]. Wnt5a is a neuronal secreted protein which is physiologically necessary for neuron excitation and pathologically sensitizes sensory neurons [54]. Li B et al. reported how gp120 through Wnt5a regulated the downstream inflammatory cytokines IL-1β, IL-1, and TNF-α secretion [8]. Yuan et al. observed that Wnt5a antagonism through Box-5 attenuated gp120-induced mechanical allodynia. Conversely, a Wnt5a agonist such as Foxy5 facilitated the allodynia. A JNK/TNF- α pathway is the downstream effector of Wnt5a, and a JNK-specific inhibitor SP600125 blocked either gp120- or Foxy5-induced allodynia as evidenced through Von Frey testing and SDH neuronal spiking evoked via mechanical stimulation of the hind paw [60]. Yuan et al. also reported that NRTIs upregulate the same Wnt5a pathway, leading to astrogliosis and the inflammatory response [71], 72]. The result showed that HIV and NRTIs form a double assault on the pain circuit through Wnt5a mediated astrogliosis, SDH sensitization and neuroinflammation. HIV-SN, signaled as cutaneous PGP9.5^+^ denervation, is a pathological mechanism for HIV-PAIN [37]. Similarly, Gp120 or Wnt5a agonism both cause PGP9.5^+^ denervation (unpublished data). Wnt5a antagonism has attenuated both gp120 and NRTI induced pain. A potential safety strategy for HIV-PAIN and HIV-SN that specifically modulates the Wnt5a pathway might be a solution. Wnt5a upregulation is found both in gp120 pain mouse model and mortem SDH, as well as in the NRTI induced process leading to chronic pain and neuroinflammation [8], 60], 113]. Further research can identify whether these combined effects constitute a true multiplicative effect. Notably, treatment of HIV-PAIN with opioids can temporally alleviate severe pain, but repeated morphine is another trigger in upregulating the Wnt5a pathway and worsens the existing allodynia and promoted astrogliosis in the SDH in mice [114]. The activation of the Wnt5a pathway leads to upregulation of pro-inflammatory cytokines IL-1β and TNF-α in mice and heightens the existing HIV-PAIN [58], 72], 114]. Thus, opioids are not a good option to treat severe neuropathic pain for PLWH. Once again, astrogliosis through the Wnt5a pathway is the key step in the pathogenesis of neuropathic pain and targeting Wnt5a might be a promising approach as a non-opioid analgesic.
Entering the CNS is a critical step in HIV-SN and early entry of HIV to the brain (1 week) facilitates the generation of viral reservoir. The primary way for HIV to initially enter the CNS is through infected monocytes by increasing monocyte chemoattractant protein-1 (CCL2) and viral replication dependent mechanisms [115]. The transmigration of infected monocytes triggers BBB disruption and short/long term neuronal and glial compromise. Disruption of this regulatory barrier allows harmful substances from the peripheral circulation to enter the CNS, thereby promoting viral entry and increasing the vulnerability to inflammation driven by immune cell activity. HIV and ART share a mechanism that facilitates increased HIV penetration into the CNS [116]. Gp120 is linked to increased permeability of the BBB in vitro and in vivo [116]. The primary mechanism of BBB disruption involves activation of neuroinflammation and increase of cytokines such as TNF-α and CCL2 [117]. Furthermore, ARTs, including Efavirenz (an NNRTI) and Dolutegravir (integrase inhibitor), cause increased BBB permeability through the disruption of tight junction structures and increased inflammatory response [32]. Thus, interference with the BBB facilitates a process by which a positive feedback loop of pathogenic molecules may enter the CNS. While this process does have some positive effect, by allowing ART to be effective in reducing the HIV viral load in the normally hidden reservoir of the CNS, at the same time, the protective function of the BBB is also diminished. Once again, a paradoxical effect of HIV and ART is observed through interactions at the BBB, further compromising neuronal/glial function and intensifying HIV-SN.
Conclusions
Despite the significant progress in managing HIV with ART, complications such as HIV-SN remain a critical concern. The interplay between HIV and ART-induced neurotoxicity exacerbates neuropathic pain through acute and chronic mechanisms, including direct viral protein toxicity, neuroinflammation, mitochondrial dysfunction, and BBB disruption. While ART has been pivotal in increasing the life expectancy of PLWH, neurotoxic effects of certain agents contribute to the burden of HIV-SN [21]. Current treatment options for HIV-SN are limited to ART modification with selection of low-risk ART and symptomatic pain management. The shared involvement of the Wnt5a signaling pathway in both HIV and ART-induced neuropathy highlights a key target for future therapeutic interventions. Furthermore, the paradoxical effects of ART on BBB integrity present challenges in balancing effective viral suppression with minimizing neurotoxicity. The use of nanomedicine as a vehicle to introduce ART to the CNS reservoir of HIV without disrupting the BBB could be effective in addressing this issue. An in vitro study from Nair et al. showed promising results of delivering ART via magneto-electro nanoparticles to the CNS [118]. After crossing the BBB, these nanoparticles are uniquely equipped to release their medication on demand to achieve higher therapeutic levels once in a latent reservoir. Other forms of nanoparticles including gold, metal oxides and core-shell formulations have also been shown to improve bioavailability with low overall toxicity [119], 120]. Utilizing novel clustered regulatory interspaced short palindromic repeat (CRISPR) is another promising strategy against HIV infection that will also benefit in nanoparticle delivery [121], 122]. Confronting the mechanisms underlying HIV-SN will be essential in reducing the long-term neurological burden associated with chronic HIV. A deeper understanding of these processes paves the way for targeted therapeutic interventions beyond symptomatic pain management. Future efforts should focus on treatments that address both the neurodegenerative effects of HIV and the adverse neurological impacts of ART, ultimately improving the quality of life for individuals living with HIV.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1UNAIDS. Global HIV Statistics UNAIDS 2024 epidemiological estimates 2025.
- 2Trickey A Sabin CA Burkholder G Crane H d’Arminio Monforte A Egger M Life expectancy after 2015 of adults with HIV on long-term antiretroviral therapy in Europe and North America: a collaborative analysis of cohort studies The Lancet HIV 202310 e 29530710.1016/S 2352-3018(23)00028-036958365 PMC 10288029 · doi ↗ · pubmed ↗
- 3Cervia LD Mc Gowan JP Weseley AJ Clinical and demographic variables related to pain in HIV-infected individuals treated with effective, combination antiretroviral therapy (c ART)Pain Med 20101149850310.1111/j.1526-4637.2010.00802.x 20210870 · doi ↗ · pubmed ↗
- 4Bruce RD Merlin J Lum PJ Ahmed E Alexander C Corbett AH 2017 HIVMA of IDSA clinical practice guideline for the management of chronic pain in patients living with HIV Clin Infect Dis 201765 e 1e 3710.1093/cid/cix 63629020263 PMC 5848240 · doi ↗ · pubmed ↗
- 5Thakur KT Boubour A Saylor D Das M Bearden DR Birbeck GL Global HIV neurology: a comprehensive review Aids 2019331638410.1097/qad.000000000000179629547440 PMC 6139090 · doi ↗ · pubmed ↗
- 6Gabbai AA Castelo A Oliveira AS HIV peripheral neuropathy Handb Clin Neurol 20131155152910.1016/b 978-0-444-52902-2.00029-123931799 · doi ↗ · pubmed ↗
- 7Benjamin LA Bryer A Emsley HC Khoo S Solomon T Connor MD HIV infection and stroke: current perspectives and future directions Lancet Neurol 2012118789010.1016/s 1474-4422(12)70205-322995692 PMC 3460367 · doi ↗ · pubmed ↗
- 8Li B Shi Y Shu J Gao J Wu P Tang SJ Wingless-type mammary tumor virus integration site family, member 5A (Wnt 5a) regulates human immunodeficiency virus type 1 (HIV-1) envelope glycoprotein 120 (gp 120)-induced expression of pro-inflammatory cytokines via the Ca 2+/calmodulin-dependent protein kinase II (Ca MKII) and c-Jun N-terminal kinase (JNK) signaling pathways J Biol Chem 201328813610910.1074/jbc.M 112.38104623539626 PMC 3650396 · doi ↗ · pubmed ↗