Association of immune relevant single nucleotide polymorphisms with ALK-positive anaplastic large cell lymphoma presentation and outcome: results of the immuno ALCL study
Petrazzuolo Adriana, Vincent Carbonnier, Fatima Domenica Elisa De Palma, Maria Perez-Lanzon, Véronique Vergé, Cyril Quivoron, Laurence Lamant, Laurence Brugieres, Charlotte Rigaud, Stéphane Ducassou, Marie-Emilie Dourthe, Marie-Cécile Le Deley, Christine Damm-Welk

TL;DR
This study explores how genetic variations in immune-related genes may influence the age of diagnosis and survival in ALK-positive anaplastic large cell lymphoma patients.
Contribution
The study identifies specific SNPs in IL10 and TLR3 genes that correlate with clinical features and survival in ALK-positive ALCL patients.
Findings
IL10 rs1800872, IL10 rs1800896, and TLR3 rs3775291 SNPs were significantly linked to age at diagnosis.
TLR3 rs3775291 was associated with progression-free survival in a recessive model.
Combining multiple genetic variations showed a trend toward post-therapeutic relapse.
Abstract
Single nucleotide polymorphisms (SNPs) of cancer-immunity relevant genes may decide the extent of tumor immunosurveillance and have clinical significance. The Immuno ALCL trial was designed to investigate whether genetic variability in 13 cancer-immunity relevant genes correlated with clinical features and outcome of anaplastic lymphoma kinase (ALK)-positive anaplastic large cell lymphoma (ALCL) patients. One hundred eighty patients were enrolled and genotyped for 14 SNPs. Age at diagnosis, progression-free survival, histological subtype and anti-ALK antibody titer data were collected. IL10 rs1800872, IL10 rs1800896 and TLR3 rs3775291 variants significantly correlated with age at diagnosis. TLR3 rs3775291 was associated with progression-free survival in a recessive model. Combination of multiple genetic variations showed a trend to associate with post-therapeutic relapse. None of the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
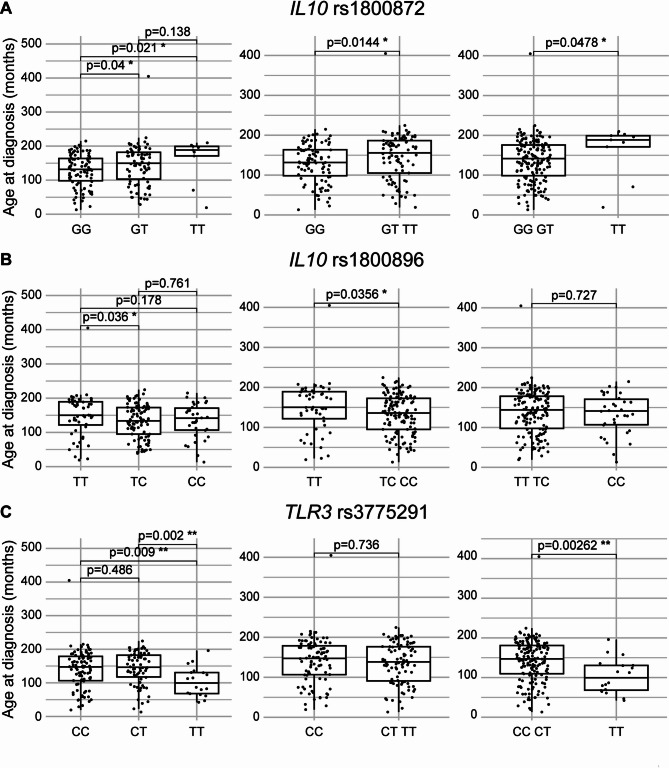 Figure 1
Figure 1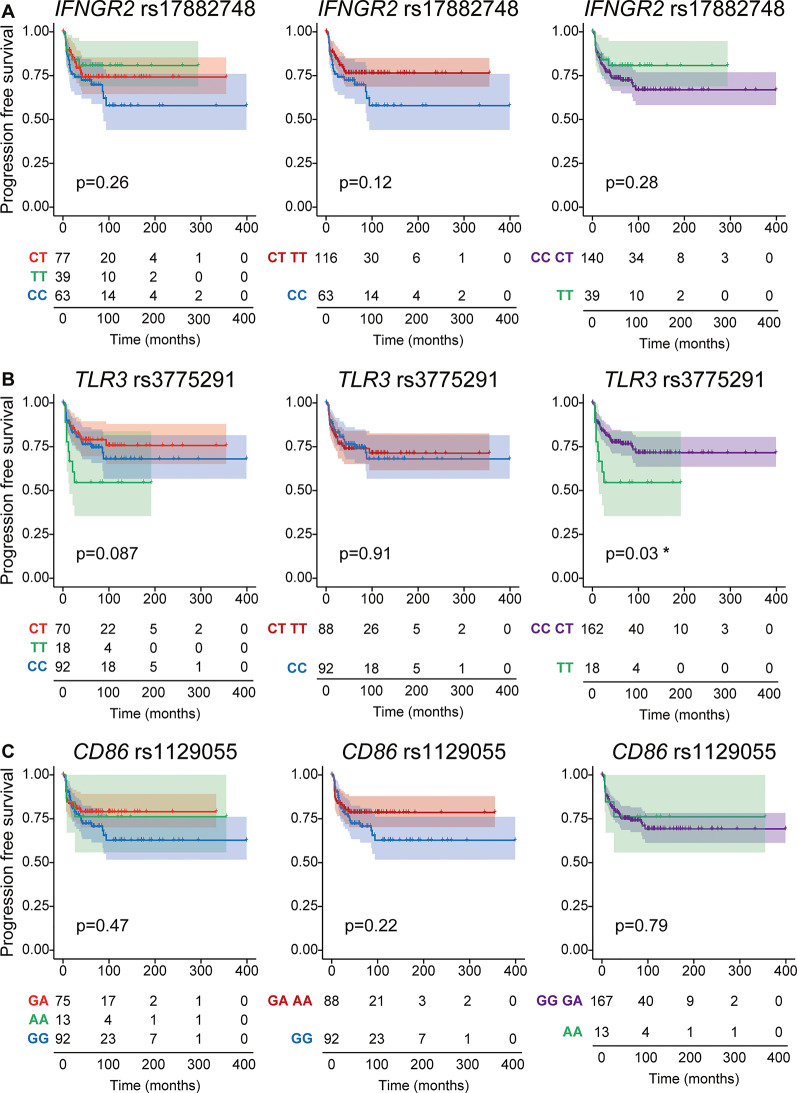 Figure 2
Figure 2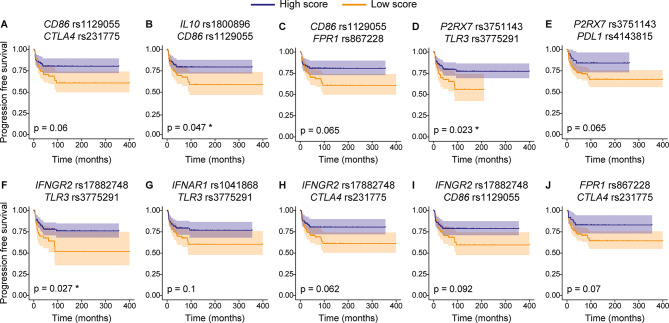 Figure 3
Figure 3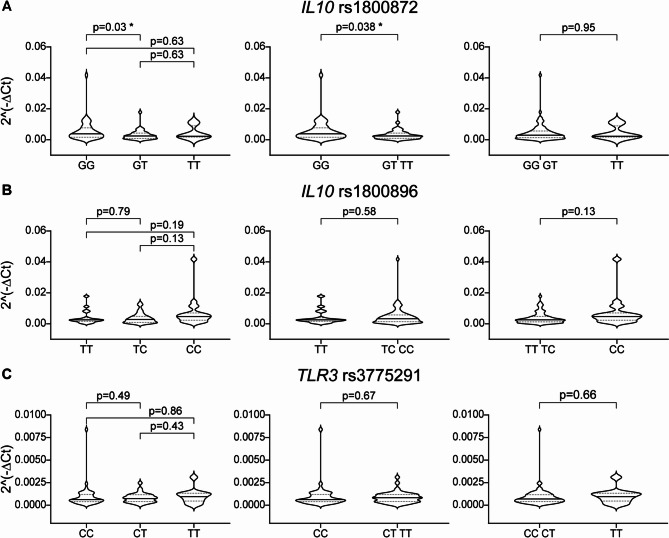 Figure 4
Figure 4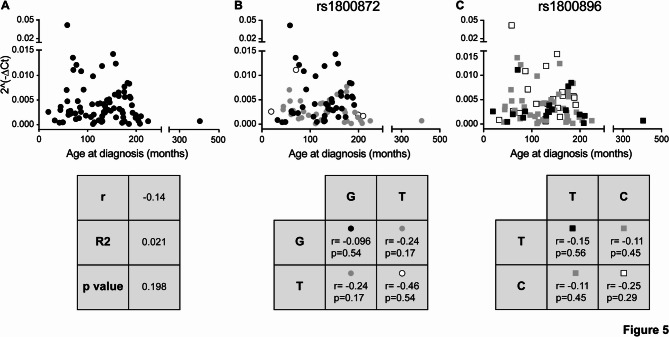 Figure 5
Figure 5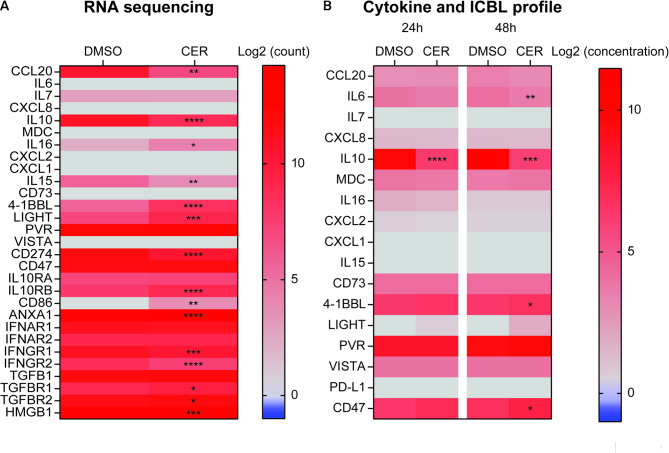 Figure 6
Figure 6- —http://dx.doi.org/10.13039/100018694HORIZON EUROPE Marie Sklodowska-Curie Actions
- —http://dx.doi.org/10.13039/501100007311Deutsche Kinderkrebsstiftung
- —http://dx.doi.org/10.13039/100018001Société Française de lutte contre les Cancers et les leucémies de l'Enfant et de l'Adolescent
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLymphoma Diagnosis and Treatment · Lung Cancer Treatments and Mutations · Immune Cell Function and Interaction
Background
Tyrosine kinase receptor anaplastic lymphoma kinase (ALK) is physiologically expressed during embryogenesis with a critical role in nervous system development. However, chromosomal rearrangements, especially translocations, cause expression of pathogenic ALK fusion proteins that drive tumor formation and maintenance. For instance, the translocation between chromosomes 2 and 5 (t(2;5)) fuses the nucleophosmin 1 (NPM1) gene with ALK [1], leading to anaplastic large cell lymphoma (ALCL) [2, 3]. The constitutive active NPM1::ALK tyrosine kinase drives tumor transformation.
Multiple lines of evidence suggest that ALK-expressing tumors are immunogenic. Accordingly, ALK-positive ALCL patients develop anti-ALK antibodies [4] and antibody titer against ALK correlate with the clinical stage of the disease and event-free survival [5, 6]. Moreover, ALK-positive ALCL patients exhibit a high frequency of circulating ALK-specific CD8^+^ T cells with an effector or memory phenotype, which indicates recent activation by antigen recognition [7]. In addition, ALK-positive ALCL patients respond to ALK-derived HLA-DRB1 (human leukocyte antigen DR beta 1 chain)-restricted peptides suggesting tumor recognition by CD4^+^ T cells [8]. These observations argue in favor of T cell- and B cell-mediated recognition of ALK-positive ALCL, which may impact tumor progression and relapse [9]. Finally, current treatments of primary or relapsed/refractory [10, 11] ALK-positive ALCL induce tumor cell death in an immunogenic fashion, thus enhancing the immune-mediated control of tumor growth [12, 13].
Genetic variability, in the form of single nucleotide polymorphisms (SNPs), of cancer-immunity relevant genes may have a prognostic or predictive value and correlate with tumor onset or risk of relapse after treatment. For instance, variants of the well-known immunosuppressive receptors PD-1 and CTLA-4, which blunt tumor immunosurveillance, correlate with the risk of developing cancer [14–17]. Likewise, SNPs in genes involved in the immune-mediated recognition of tumor cells upon their immunogenic death, are associated with disease onset and treatment efficacy. Our group demonstrated that the loss-of-function variant rs867228 of FPR1 is associated with precocious onset of breast, colorectal, esophageal and head & neck carcinomas [18]. Furthermore, SNP rs4986790 of TLR4 and rs3751143 of P2RX7 correlate with the frequency of metastases after surgery in women with breast cancer [19, 20]. Since the progression of ALK-positive ALCL is influenced by therapy-independent and therapy-induced anti-tumor immune response, we investigated whether the genotype of 14 cancer-immunity relevant SNPs might correlate with lymphoma presentation and recurrence, as well as with histological features of ALK-positive ALCL.
Materials and methods
Study population
Patients from both French and German centers were included in the study. The French cohort consisted of individuals over 3 years of age enrolled in prospective therapeutic trials for ALK-positive ALCL between 1975 and 2020 [21, 22]. Clinical and biological data were collected from the ALCL99, HM91 and COPAD-IGR databases. Written informed consent was obtained from patients or legal guardians, and assent was provided by minors when appropriate. The German cohort included patients treated with BFM-type regimens in the NHL-BFM 95, ALCL99 and NHL-BFM Registry 2012 studies. All participants had consented to an ethically approved protocol investigating immune responses to ALK, which included genetic testing.
Study design and procedure
For the French group (n = 150), eligible patients were identified in the national ALCL database and confirmed by investigators at each participating Société Française de lutte contre les Cancers et leucémies de l’Enfant et de l’adolescent (SFCE) center. Investigators updated clinical data, verified availability of stored sera, obtained consent from surviving patients and organized blood collection. Samples were processed at Gustave Roussy for peripheral blood mononuclear cells (PBMCs) and serum isolation. The study was approved by relevant ethics committees and national authorities and conducted in accordance with the Declaration of Helsinki. The median time from diagnosis to blood sampling was 2.1 years.
For the German group (n = 30), blood samples were obtained from patients aged 14 years or older in complete remission. DNA was extracted from PBMCs after double pseudonymization. Anti-ALK antibodies were assessed at diagnosis. Clinical and biological data were provided through the ALCL99 or NHL-BFM registry. ClinicalTrials.gov ID NCT02902874.
DNA extraction and genotyping
DNA was extracted from PBMCs using the DNeasy Blood & Tissue Kits (Qiagen, Hilden, Germany) following manufacturer’s instruction. SNP genotyping was performed with TaqMan assays (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and TaqMan genotyping Master Mix (Thermo Fisher Scientific) on the StepOnePlus Real-Time PCR System (Applied Biosystems, Waltham, Massachusetts, USA). Cycling conditions were 60 °C for 30 s, 95 °C for 10 min and 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Allelic discrimination was based on FAM/VIC fluorescence (Table 1).
Table 1. List of SNPs IDs and TaqMan assay IDsGeneSNP IDAssay IDCD274rs4143815C__31941235_10CD86rs1129055C___7504226_10CTLA4rs231775C___2415786_20ECE1rs1076669C___2464666_30FPR1rs867228C___3266374_1_IFNAR1rs1041868C___1841019_10IFNGR2rs17882748C__61106388_10IL10rs1800872C___1747363_10IL10rs1800896C___1747360_10PDCD1rs2227981C__57931286_20TGFB1rs1800469C___8708473_10P2RX7rs3751143C__27495274_10TLR3rs3775291C___1731425_10TLR4rs4986790C__11722238_20
Cell culture and treatment
Human NPM1::ALK-positive anaplastic large cell lymphoma (ALCL) SU-DHL-1 cell line was purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany) and cultured in RPMI 1640 supplemented with 100 units/mL of penicillin, 100 µg/mL of streptomycin (Pen/Strep) and 10% fetal bovine serum (FBS). Cells were grown in standard conditions (37 °C and 5% CO2) and routinely checked for mycoplasma contamination. All media and supplements were purchased from Gibco (Waltham, Massachusetts, USA). ALCL cells were treated with the ALK inhibitor ceritinib (CER; Selleck Chemicals, Houston, Texas, USA) at 50 nM for 24–48 h.
RNA extraction and RT-qPCR
RNA was extracted from PBMCs using the RNeasy plus mini kit (Qiagen) following manufacturer’s instruction. Reverse transcription to cDNA was performed using SuperScript™ IV VILO™ Master Mix (Thermo Fisher Scientific). TLR3, IL10 or GAPDH were amplified with specific TaqMan Gene Expression assays (Thermo Fisher Scientific) using the TaqMan Fast Advanced Master Mix (Thermo Fisher Scientific) on the StepOnePlus Real-Time PCR System (Applied Biosystems). Relative expression levels were calculated and 2^(−ΔCt) transformation was used for analysis.
RNA sequencing
RNA was extracted from cell pellets using the RNeasy plus mini kit (Qiagen) and RNA quality (RNA integrity number or RIN) was determined on the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, California, USA) prior to library preparation with TruSeq Stranded mRNA kits (Illumina). Libraries were then quantified by using the KAPA Library Quantification Kit for Illumina Libraries (KapaBiosystems, Wilmington, Massachusetts, USA), profiled by using the DNA High Sensitivity LabChip kit and finally sequenced on an Illumina Nextseq 500. Data processing used AOZAN software (ENS Paris) for demultiplexing and controlling quality of raw data, STAR algorithm (version 2.5.2b) and Picard tools (version 2.8.1) for alignment and Featurecount (version Rsubread 1.24.1) for read counts. Raw data have been made publicly available on the repository ArrayExpress (accession number E-MTAB-15866).
Cytokines and immune checkpoint ligands quantification
After treatment, supernatants were collected, and 20 cytokines and immune checkpoint ligands were quantified using Human Procarta Plex kits (Thermo Fisher Scientific) by following the manufacturer’s instructions.
Detection of anti-ALK antibodies
Detection of anti-ALK antibodies in patients’ sera was performed as previously described by Ait-Tahar and colleagues [5]. Briefly, COS-1 cells (DSMZ) were transiently transfected with ALK-encoding plasmids, incubated with patient’s serum and stained with a rabbit anti-human IgG-HRP. Titers were defined as the highest serum dilution showing a positive signal [4] with 1/750 as the cutoff [6].
Statistical analysis
All clinical data were pseudonymized prior to analysis. Age at diagnosis and progression-free survival (PFS) were derived from registry data. Associations with SNP genotypes were tested by using Mann-Whitney and log-rank statistical tests. A score-based system was used to assess the correlation between combinations of SNP and PFS. A score of 0, 1 or 2 was assigned to each SNP genotype based on the association with PFS. Lower scores were assigned to genotypes associated with higher risk of relapsing, higher scores to genotypes that correlated with lower risk of relapsing. Patients were grouped in two classes (low or high score) and Kaplan-Meier estimates were used to plot PFS according to patient score. Additional analyses included Fisher’s exact test, Chi-square, student’s t test and Bootstrap t-test p-value adjusted by the Benjamini-Hochberg procedure, with 10,000 permutations. R and GraphPad Prism v9 were used for analysis.
Results
Genotyping
One hundred eighty patients diagnosed with ALK-positive ALCL were enrolled in this study. Patients were genotyped for 14 cancer-immunity relevant SNPs, considered as relevant based on our previous works and literature search [14, 15, 18–20, 23–31]. Frequency in the global population and clinical relevance of SNPs are reported in Table 2 (source www.ncbi.nlm.nih.gov/snp and www.ncbi.nlm.nih.gov/clinvar). The frequency in the study population as well as clinical data according to SNP genotype are presented in Table 3.
Table 2SNP frequency and characteristicsClinical impact (Clinvar)Allele frequency in global populationMolecular consequencesImpactDisease CD274 rs4143815G = 0.73823Non coding transcript variantNot reported in ClinVarC = 0.26177 CD86 rs1129055G = 0.726306Missense variant (Ala310Thr)BenignA = 0.273694 CTLA4 rs231775A = 0.617783Missense variant (Thr17Ala)Risk factorHashimoto thyroiditis [32]; Thyroid-associated orbitopathy [33]; Systemic lupus erythematosus [34, 35]; Celiac disease [36, 37]; Type 1 and 2 Diabetes [38–41]G = 0.382217BenignAutoimmune lymphoproliferative syndrome due to CTLA4 haploinsuffiency ECE1 rs1076669G = 0.935386Missense variant (Thr341Ile)BenignA = 0.064614 FPR1 rs867228T = 0.201138Missense variant (Glu346Ala)BenignGingival disorderG = 0.798862 IFNAR1 rs1041868G = 0.812593Intron variantNot reported in ClinVarA = 0.187407 IFNGR2 rs17882748T = 0.417765 Prime UTR variantBenignC = 0.58224 IL10 rs1800872T = 0.2742622KB upstream variantRisk factorHIV infection [42]G = 0.725738BenignInflammatory bowel diseaseProtectiveGraft-versus-host disease [43]IL10 rs1800896T = 0.5534502KB upstream variantNot reported in ClinVarC = 0.446550 PDCD1 rs2227981A = 0.42806Synonymous variantBenignG = 0.57194 P2RX7 rs3751143A = 0.815741Missense variant (Glu496Ala)Not reported in ClinVarC = 0.184259 TGFB1 rs1800469A = 0.3194842KB upstream variantBenignJoubert syndrome, Meckel-Gruber syndromeG = 0.680516 TLR3 rs3775291C = 0.723658Missense variant (Leu412Phe)BenignSusceptibility to Herpes simplex encephalitis [44]T = 0.276342ProtectiveSusceptibility to HIV infection [45] TLR4 rs4986790A = 0.941860Missense variantAsp299GlyProtectivePericementitis [46, 47]G = 0.058140
Table 3. Study population genotypes and clinical dataGenotype frequencyProgressionMedian age at diagnosisn/tot(%)YES n/tot(%)NOn/tot(%)Chi-square P valueMedianCI 95% CD274 rs4143815GG102/180(56.7)26/102 (25.5)76/102 (74.5)0.5268141130–156GC59/180(32.8)17/59 (28.8)42/59 (71.2)141116–154CC19/180(10.6)3/19 (15.8)16/19 (84.2)172122–183 CD86 rs1129055GG92/180(51.1)28/92 (30.4)64/92 (69.6)0.2997136124–152GA75/180(41.7)15/75 (20)60/75 (80)147135–163AA13/180(7.22)3/13 (23.1)10/13 (76.9)145120–166 CTLA4 rs231775AA85/180(47.2)23/85 (27.1)62/85 (72.9)0.5166145130–154AG75/180(41.7)20/75 (26.7)55/75 (73.3)137122–155GG20/180(11.1)3/20 (15)17/20 (85)158106–169 ECE1 rs867228GG160/180(88.9)40/160 (25)120/160 (75)0.4833143130–153GA20/180(11.1)6/20 (30)14/20 (70)138122–174 FPR1 rs867228GG104/180(57.8)27/104 (26)77/104 (74)nc143134–163GT70/180(38.9)18/70 (25.7)52/70 (74.3)145128–155TT6/180(3.33)1/6 (16.7)5/6 (83.3)10575.5–140 IFNAR1 rs1041868GG117/180(65)33/117(28.2)84/117 (71.8)0.4602142129–147GA55/180(30.6)12/55(21.8)43/55 (78.2)150124–174AA8/180(4.44)1/8(12.5)7/8 (87.5)122105–154 IFNGR2 rs17882748CC63/179(35.2)21/63 (33.3)42/63 (66.7)0.1857149130–158CT77/179(43)18/77(23.4)59/77 (76.6)142129–163TT39/179(21.8)7/39 (17.9)32/39 (82.1)135106–161 IL10 rs1800872GG91/180(50.6)25/91 (27.5)66/91 (72.5)0.8330131123–142GT80/180(44.4)19/80 (23.8)61/80 (76.2)150135–171TT9/180(5)2/9 (22.2)7/9 (77.8)18870–199 IL10 rs1800896TT52/180(28.9)14/52 (26.9)38/52 (73.1)0.9396150139–178TC94/180(52.2)24/94 (25.5)70/94 (74.5)133122–153CC34/180(18.9)8/34 (23.5)26/34 (76.5)141120–160 PDCD1 rs2227981GG48/180(26.7)14/48 (29.2)34/48 (70.8)0.6196130112–158GA101/180 (56.1)26/101 (25.7)75/101 (74.3)145135–157AA31/180(17.2)6/31 (19.4)25/31 (80.6)128112–173 P2RX7 rs3751143AA105/180(58.3)30/105 (28.6)75/105 (71.4)nc142129–155AC72/180(40)16/72 (22.2)56/72 (77.8)146131–163CC3/180(1.67)0/3 (0)3/3 (100)9179–187 TGFB1 rs1800469GG82/180(45.6)23/82 (28)59/82 (72)0.7808148128–161GA72/180(40)17/72 (23.6)55/72 (76.4)142132–161AA26/180(14.4)6/26 (23.1)20/26 (76.9)13898–163 TLR3 rs3775291CC92/180(51.1)23/92 (25)69/92 (75)0.1342146130–161CT70/180(38.9)15/70 (21.4)55/70 (78.6)146134–168TT18/180(10)8/18 (44.4)10/18 (55.6)9968–130 TLR4 rs4986790AA165/180(91.7)43/165 (26.1)122/165 (73.9)0.5152142129–150AG15/180(8.33)3/15 (20)12/15 (80)14161–163nc: criteria for Chi-square test were not met
Association of polymorphisms with age at diagnosis
Patient age at diagnosis was evaluated according to the genotype at fourteen SNP loci and three out of 14 SNPs were significantly associated with age at diagnosis: IL10 rs1800872, IL10 rs1800896 and TLR3 rs3775291 (Fig. 1). The presence of at least one T nucleotide at rs1800872 locus associated with late disease onset (p = 0.0144). Median age at diagnosis of patients with the GG genotype was 131 months (CI 95% 123–142), whereas it increased to 150 (CI 95% 135–171) and 188 months (CI 95% 70–199) for patients bearing the GT and TT genotypes, respectively. Rs1800896 was also significantly associated with age at diagnosis. Notably, patients bearing at least one C presented with earlier onset than patients with TT genotype (p = 0.0356). Patients bearing SNP variant TT were diagnosed at 150 months (CI 95% 139–178) of age, whereas median diagnosis occurred at 133 (CI 95% 122–153) and 141 months (CI 95% 120–160) in patients bearing TC and CC variants, respectively. Finally, SNP rs3775291 of TLR3 gene also associated with ALCL occurrence. The presence of a T nucleotide in homozygosity was associated with early diagnosis (99 months CI 95% 68–130). Diagnosis was significantly accelerated by 47 months as compared to the CT genotype (146 months CI 95% 134–168, p = 0.002) and as compared to the more frequent CC genotype (146 months CI 95% 130–161, p = 0.009).
Fig. 1. Age at diagnosis according to IL10 and TLR3 SNPs. Boxplots displaying median and interquartile ranges of ALK-positive ALCL patients age at diagnosis according to the genotype of selected SNPs. rs1800872 is shown in (A), rs1800896 in (B) and rs3775291 in (C). Dominant or recessive behavior of the variant allele was tested by grouping patients bearing at least one variant or reference allele (middle and right panels). Mann-Whitney test was used to calculate statistical significance, expressed as p values and *. *p < 0.05, **p < 0.01
Association of polymorphisms with PFS
At the time of analysis 46 out of 180 patients enrolled in this study experienced a relapse event. Association of genetic polymorphisms with PFS suggested trends for IFNGR2 rs17882748, TLR3 rs3775291 and CD86 rs1129055 to segregate patients with high risk from those who had low risk of relapsing after therapy (Fig. 2). TLR3 rs3775291 seemed to associate with the risk of tumor recurrence in a recessive model. Patients bearing the T nucleotide in homozygosity had a statistically significantly higher risk of relapsing after therapy when compared to those having CC and CT at rs3775291 locus (TT vs. CT + CC p = 0.03). No other association between single SNPs and PFS was detected (Figure S1). We next evaluated whether the combination of multiple genetic variations correlated with PFS. Thus, we generated a score for each patient based on his or her polymorphism profile. The presence of alleles associated with higher PFS received higher score. Of all possible SNP combinations, ten of them tended to stratify patients in two groups with different risks of disease recurrence after chemotherapy (Fig. 3). Interestingly, the combination of SNP rs1129055 in CD86 and rs231775 in CTLA4 appeared to correlate with PFS as did the combination of rs1129055 and rs1800896 in IL10. Noteworthy, rs1129055 of CD86 recurred in three different combinations.
Fig. 2. Progression-free survival of ALK-positive ALCL-patients according to selected SNP genotypes. Kaplan-Meier plots displaying progression-free survival of patients with ALK-positive ALCL after chemotherapy. Patients were categorized according to selected SNP genotypes. IFNGR2 rs17882748 is shown in (A), TLR3 rs3775291 in (B) and CD86 rs1129055 in (C). Dominant or recessive behavior of the variant allele was tested by grouping patients bearing at least one variant or reference allele (middle and right panels). Observations of patients alive without failure were censored at the time of their last follow-up and indicated by a cross on the curve at the censoring time. Log-rank test was used to calculate statistical significance, expressed as p values and *. *p < 0.05. Numbers at the bottom of the figure are number at risk
Fig. 3. Progression-free survival of ALK-positive ALCL-patients according to selected combination of SNPs. Kaplan-Meier plots displaying progression-free survival of patients with ALK-positive ALCL after chemotherapy. Patients were categorized according to selected combination of SNPs after scoring, rs1129055 and rs231775 (A), rs1800896 and rs1129055 (B), rs1129055 and rs867228 (C), rs3751143 and rs3775291 (D), rs3751143 and rs4143815 (E), rs17882748 and rs3775291 (F), rs1041868 and rs3775291 (G), rs17882748 and rs231775 (H), rs17882748 and rs1129055 (I), rs867228 and rs231775 (J). Observations of patients alive without failure were censored at the time of their last follow-up and indicated by a cross on the curve at the censoring time. Log-rank test was used to calculate statistical significance, expressed as p values and *. *p < 0.05.
Association of polymorphisms with histologic subtype and anti-ALK antibody titers
The World Health Organization (WHO) recognizes five different subtypes of ALK-positive ALCL: common pattern, small cell pattern, lymphohistiocytic pattern, Hodgkin’s-like pattern and composite pattern. The small cell pattern (SC) and lymphohistiocytic subtype (LH) are independent risk factors determining a higher risk of relapsing compared to other subtypes [48, 49].
Reference histology was available for 96 patients. Thirty patients had ALCL with SC or LH subtypes and 66 ALCL with other patterns. Genotype frequency of the 14 cancer-immunity relevant SNPs according to the histological subtype is shown in Table 4. Genotypes were similarly distributed among SC/LH and other patterns (Table 4).
Table 4. Association of SNP genotype with histologic subtype of ALK-positive ALCL and circulating anti-ALK antibody levelHistological subtypeAnti-ALK antibody levelOther subtypesn/tot(%)SC/LHn/tot(%)Fisher’s Exact Testp valueLow titern/tot(%)High titern/tot(%)Fisher’s Exact Testp value CD274 rs4143815GG39/96(40.6)17/96(17.7)0.593711/80(13.8)39/80(48.8)0.4743GC19/96(19.8)11/96(11.5)7/80(8.7)12/80(15)CC8/96(8.3)2/96(2.1)3/80(3.7)8/80(10) CD86 rs1129055GG33/96(34.4)19/96(19.8)0.401311/80(13.8)29/80(36.2)0.6412GA29/96(30.2)9/96(9.4)8/80(10)27/80(33.8)AA4/96(4.2)2/96(2.1)2/80(2.5)3/80(3.7) CTLA4 rs231775AA31/96(32.3)15/96(15.6)0.825112/80(15)27/80(33.8)0.1604AG32/96(33.3)13/96(13.5)9/80(11.2)23/80(28.8)GG3/96(3.1)2/96(2.1)0/80(0)9/80(11.2) ECE1 rs867228GG59/96(61.5)29/96(30.2)0.428318/80(22.5)50/80(62.5)1GA7/96(7.3)1/96(1)3/80(3.8)9/80(11.2) FPR1 rs867228GG39/96(40.6)21/96(21.9)0.626711/80(13.8)37/80(46.2)0.2784GT24/96(25)8/96(8.3)10/80(12.5)18/80(22.5)TT3/96(3.1)1/96(1)0/80(0)4/80(5) IFNAR1 rs1041868GG44/96(45.8)21/96(21.9)0.564216/80(20)37/80(46.2)0.5277GA18/96(18.7)9/96(9.4)5/80(6.3)20/80(25)AA4/96(4.2)0/960/80(0)2/80(2.5) IFNGR2 rs17882748CC21/96(21.9)12/96(12.5)0.686610/79(12.7)17/79(21.5)0.2863CT30/96(31.3)13/96(13.5)8/79(10.1)26/79(32.9)TT15/96(15.6)5/96(5.2)3/79(3.8)15/79(19) IL10 rs1800872GG30/96(31.2)16/96(16.7)0.457312/80(15)30/80(37.5)0.4992GT32/96(33.3)14/96(14.6)9/80(11.2)24/80(30)TT4/96(4.2)0/960/80(0)5/80(6.3)IL10 rs1800896TT19/96(19.8)6/96(6.2)0.69018/80(10)17/80(21.2)0.6232TC34/96(35.4)18/96(18.8)11/80(13.8)31/80(38.8)CC13/96(13.5)6/96(6.2)2/80(2.5)11/80(13.7) PDCD1 rs2227981GG12/96(12.5)9/96(9.4)0.21447/80(8.8)16/80(20)0.9425GA45/96(46.9)15/96(15.6)10/80(12.5)31/80(38.7)AA9/96(9.4)6/96(6.2)4/80(5)12/80(15) P2RX7 rs3751143AA34/96(35.4)21/96(21.9)0.23149/80(11.2)37/80(46.2)0.2049AC30/96(31.2)9/96(9.4)12/80(15)21/80(26.3)CC2/96(2.1)0/960/801/80(1.3) TGFB1 rs1800469GG30/96(31.3)15/96(15.6)0.66229/80(11.2)28/80(35)0.948GA28/96(29.2)10/96(10.4)9/80(11.3)22/80(27.5)AA8/96(8.3)5/96(5.2)3/80(3.8)9/80(11.2) TLR3 rs3775291CC31/96(32.3)16/96(16.6)0.730112/80(15)31/80(38.7)0.2584CT28/96(29.2)10/96(10.4)6/80(7.5)25/80(31.2)TT7/96(7.3)4/96(4.2)3/80(3.8)3/80(3.8) TLR4 rs4986790AA60/96(62.5)27/96(28.1)121/80(26.2)54/80(67.5)0.3184AG6/96(6.3)3/96(3.1)0/805/80(6.3)
Anti-ALK antibody titer in serum or plasma is a surrogate marker of the strength of a specific anti-tumor immune response orchestrated by T lymphocytes and mediated by B cells. Low anti-ALK antibody titer at diagnosis have been shown to correlate with high relapse risk [5, 6]. We investigated whether SNPs of the 13 cancer-immunity relevant genes would associate with the anti-ALK antibody titer of a subset of patients (n = 80). None of the SNPs tested correlated to the titer when grouped as previously published (Table 4).
Association of polymorphisms with TLR3 and IL10 gene expression
Single nucleotide variations may affect gene function or expression depending on their location. Upstream variants alter the promoter sequences of genes, thus regulating their transcription and the abundance of mRNAs and proteins. Alternatively, SNPs located in exons may modify the protein sequence, thus altering the structure, stability and function. We wondered whether TLR3 or IL10 gene expression would be affected by rs3775291 or rs1800872/rs1800896 genotypes, respectively. To address this question, we quantified TLR3 and IL10 transcripts in PBMCs isolated from 85 patients and bearing the three alternative variants of the SNPs. The rs3775291 genotype had no impact on TLR3 RNA levels. However, both rs1800872 and rs1800896 modestly correlated with IL10 gene expression, as expected by SNP location in the promoter region (Table 2). The presence of at least one T at rs1800872 was associated with a lower median of IL10 expression as compared to the GG genotype (GT TT vs. GG fold change = 0.63, p = 0.038). Similarly, patients bearing the CC allele at rs1800896 showed a trend for higher IL10 expression (CC vs. TT TC fold change = 1.8, p = 0.13) (Fig. 4). Linear correlation between age at diagnosis and IL10 mRNA levels did not reach statistical significance due to the limited number of samples (Fig. 5). However, even if not statistically significant, IL10 expression tended to negatively correlate with age at diagnosis for patients heterozygous at rs1800872 site (Fig. 5). The subgroup of heterozygous patients experiencing later diagnosis were having lower level of IL-10 mRNA.
Fig. 4IL10 and TLR3 expression levels according to rs1800872, rs1800896 and rs3775291 genotypes. IL10 and TLR3 mRNA levels in circulating PBMCs of ALK-positive ALCL patients (n = 85) were assessed using qRT-PCR. Relative expression, as 2^(−ΔCt), is presented according to rs1800872 (A), rs1800896 (B) or rs3775291 (C) genotypes. Statistical significance was calculated using the Student’s t-test with Welch’s correction and expressed as p values and *. *p < 0.05, **p < 0.01
Fig. 5IL10 mRNA levels, expressed as 2^(−ΔCt), of ALK-positive ALCL patients (n = 82) are shown against patients age at diagnosis, expressed in months (A). Correlation between IL10 expression level and age at diagnosis according to SNP genotype of rs1800872 (B) and rs1800896 (C). Pearson’s correlation coefficients and statistical significance are shown in the box below the respective graph
Expression profile of immune-related genes by ALK-positive ALCL tumor
Next, we characterized the expression of immune-relevant genes in an ALK-positive ALCL cell line, namely SU-DHL-1. We took advantage of the treatment with ceritinib, second generation ALK inhibitor, to reveal any ALK-dependent effect. For that purpose, we sequenced SU-DHL-1 transcriptome upon treatment with ceritinib and measured secretion of a panel of 20 cytokines and immune-related ligands after ALK inhibition (Fig. 6). Notably, SU-DHL-1 expressed many immune-related transcripts in an ALK-dependent manner, because expression significantly decreased after ceritinib treatment (CCL20, IL10, IL15, CD274 or IFNGR). On the contrary, ALK kinase activity repressed the expression of CD86 and the B subunit of IL10 receptor (IL10RB), because evident expression was found after ALK inhibition. Expression at the mRNA level paralleled with protein expression and/or secretion for some of the cytokines and immune-related ligands analyzed, namely IL10 (Fig. 6).
Fig. 6. Transcriptome and secretome of ALK-positive ALCL cell line SU-DHL-1. Transcriptomic profile of selected mRNAs of human NPM1::ALK-positive SU-DHL-1 cells treated with the ALK inhibitor ceritinib (50nM) for 24 h. The expression level of immune-relevant transcripts of ceritinib-treated cells were compared to vehicle-treated cells and fold changes (in logarithmic scale) are depicted (A). Secretion profile of selected cytokines and immune-checkpoint ligands of human NPM1::ALK-positive SU-DHL-1 cells treated with the ALK inhibitor ceritinib (50nM) for 24–48 h. Cytokine and immune checkpoint ligands levels of ceritinib-treated cells were compared to vehicle-treated cells and fold changes (in logarithmic scale) are depicted (B). Statistical significance was calculated using the multiple Student’s t-test. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001
It appears that ALK-positive ALCL tumor, or at least a representative cell line, presents many receptors and/or ligands, which participate in the anti-tumor immune response and possibly skew it toward its growth.
Discussion
Many lines of evidence indicate that the anti-tumor immune response, which develops naturally or after immunogenic therapy, contributes to eradicate ALK-positive ALCL [4, 5, 7–9, 50]. With the aim of deepening the knowledge of immune-based mechanisms that might influence tumor onset and relapse in ALK-positive ALCL patients, we assessed 14 genetic variants of 13 genes potentially involved in the anti-tumor immune response in 180 patients. First, we found that age at diagnosis differed among patients carrying genetic variants of TLR3 and IL10, suggesting a role of these pathways in the spontaneous, therapy-independent anti-tumor immune response. TLR3 contributes to the activation of the innate immune response [51, 52] and the substitution C >T at rs3775291 leads to an amino acid change, which reduces TLR3 dimerization and RNA binding capacity, thus compromising downstream signaling [53]. Interestingly, ALK-positive ALCL patients bearing TT at rs3775291 were diagnosed at a comparatively earlier age. Notably, TLR3 has also a critical role in immunogenic therapy-induced immune response in breast cancer patients [23, 24]. ALCL patients included in our study were treated with a chemotherapy regimen, which included doxorubicin, a very well-known immunogenic cell death-inducing agent [10, 54]. TLR3 genetic polymorphism also showed a trend to segregate patients with high risk from those who had low risk of relapsing after therapy, and the presence of the loss-of function allele (T) in homozygosity, which occurs in 8% of the global population, tended to correlate with a higher risk of tumor recurrence.
Our analysis also suggested with caution that malignant disease was diagnosed later in patients with reduced expression of IL10 mRNA. Noteworthy, patients bearing at least one T at rs1800872 (which occurs in 29% of the general population), were diagnosed relatively late and presented with lower IL10 mRNA expression. Although transcript levels do not necessarily reflect protein levels and further confirmation in a larger cohort of patients is needed, it is tempting to speculate that high IL10 levels, associated with unfavorable SNPs, suppressed the spontaneous anti-tumor immune response and hence accelerated the development of ALK-positive ALCL. Notably, IL10 may also have a direct effect on ALK-positive ALCL tumor cells because the ALCL cell line, SU-DHL1, expresses IL10 receptor subunits A and B in an ALK-dependent fashion (Fig. 6). In these cells, activation of IL10 receptor signaling triggers STAT3 phosphorylation, which in turn promotes tumor cell survival and proliferation. Moreover, transfection-enforced overexpression of IL10RA confers resistance to ALK inhibitors [55].
Finally, we assessed whether certain combinations of SNPs were associated with patient prognosis. We uncovered several combinations that showed a trend to predict prognosis, although such effects must be confirmed in a larger cohort of patients. However, the combination of two SNPs, namely rs1129055 of CD86 gene and rs231775 of the CTLA4 gene caught our attention because CD86 and CTLA4 engage in a receptor-ligand interaction that inhibit anti-tumor immune response [56]. The G >A transition in CD86 gene (rs1129055), which occurs in 27% of the global population (at least at one allele), introduces a phosphorylation site in the cytoplasmic region of CD86, which alters its downstream signals [57]. Patients with kidney transplantation harboring AA genotype exhibited a reduced risk of acute rejection, suggesting a reduced level of immune activation [58]. Similarly, the A>G transition at rs231775 affecting CTLA4 causes an amino acid substitution in CTLA4 protein that reduces the expression levels of the receptor. Accordingly, the rs231775 GG genotype, occurring in 13.8% of the general population, is associated with higher T cell activation and proliferation and, in fact, is more frequent in patients with autoimmune disorders, such as rheumatoid arthritis and Hashimoto thyroiditis [59–62]. Our results suggested that the simultaneous presence of GG at rs1129055 and at least one A at rs231775 tended to correlate with a higher risk of post-therapeutic relapse. These results seem to be, at least in part, in contradiction with the assumption that higher T cell activation corresponds to successful tumor control. However, anti-tumor immune response becomes efficient because of the fine tuning of co-stimulatory and inhibitory signals. Of note, excess of inhibitory signals may favor tumor escape. Moreover, when inflammatory or co-stimulatory signals prevail, tumor cell proliferation may predominate, or autoimmunity arise as unwanted side effect. The discrepancy between trends shown by our results and known effect of rs1129055 on immune system activation may be also explained by the expression of CD86 by ALK-positive ALCL tumor cells. SU-DHL-1 expressed CD86, at the mRNA level (Fig. 6), and CD86 of tumor origin might intervene to alter the thin equilibrium that decides T cell activation threshold.
Limitation of the study
We believe that our findings reinforce the clinical relevance of immunosurveillance in the ultimate control of ALK-positive ALCL and support a potential contribution of the IL10 and TLR3 pathways to its pathogenesis. Nevertheless, certain limitations should be acknowledged. First, because ALCL is a rare disease with an incidence of approximately 1.2 cases per million persons per year, our patient cohort was small, and the study may be underpowered to detect the effect of individual single-nucleotide variants on disease presentation and progression. Therefore, validation in larger patient cohorts is essential before our observations can be considered definitive. Moreover, while our data suggest a role for the IL10 and TLR3 pathways in ALK-positive ALCL pathogenesis, these associations do not establish causality. Further mechanistic studies are required to substantiate the potential pro-ALCL effects of IL10 and TLR3.
Conclusion
To sum up, despite its limitations, our study further supports the importance of immunosurveillance in the ultimate control of ALK-positive ALCL. Consistent with this notion, genetic variants influencing the threshold of immune activation were found to be associated with patient age at diagnosis and risk of relapse, suggesting that the IL10 and TLR3 pathways contribute to the pathogenesis of ALK-positive ALCL.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplementary Material 1
Supplementary Material 2
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Leventaki V, Shaw T, Pounds S, Cao X, Ma J, Palacios G, et al. Comprehensive genomic analysis reveals molecular heterogeneity in pediatric ALK-positive anaplastic large cell lymphoma [Internet]. 2024. Available from: https://www.researchsquare.com/article/rs-4145750/v 1.10.1038/s 41375-024-02468-4PMC 1277179039592809 · doi ↗ · pubmed ↗
- 2Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer. 2013.10.1038/nrc 358024060861 · doi ↗ · pubmed ↗
- 3Pulford K, Falini B, Banham AH, Codrington D, Roberton H, Hatton C, et al. Immune response to the ALK oncogenic tyrosine kinase in patients with anaplastic large-cell lymphoma. Blood. 2000.10942417 · pubmed ↗
- 4Ait-Tahar K, Damm-Welk C, Burkhardt B, Zimmermann M, Klapper W, Reiter A, et al. Correlation of the autoantibody response to the ALK oncoantigen in pediatric anaplastic lymphoma kinase-positive anaplastic large cell lymphoma with tumor dissemination and relapse risk. Blood. 2010.10.1182/blood-2009-11-25189220185586 · doi ↗ · pubmed ↗
- 5Passoni L, Gallo B, Biganzoli E, Stefanoni R, Massimino M, Di Nicola M, et al. In vivo T-cell immune response against anaplastic lymphoma kinase in patients with anaplastic large cell lymphomas. Haematologica. 2006.16434370 · pubmed ↗
- 6Prokoph N, Larose H, Lim MS, Burke GAA, Turner SD. Treatment options for paediatric anaplastic large cell lymphoma (ALCL): current standard and beyond. Cancers (Basel). 2018;10(4).10.3390/cancers 10040099 PMC 592335429601554 · doi ↗ · pubmed ↗
- 7Liu P, Zhao L, Pol J, Levesque S, Petrazzuolo A, Pfirschke C, et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat Commun. 2019.10.1038/s 41467-019-09415-3PMC 644509630940805 · doi ↗ · pubmed ↗
- 8Vacchelli E, Ma Y, Baracco EE, Sistigu A, Enot DP, Pietrocola F, et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science (1979). 2015.10.1126/science.aad 077926516201 · doi ↗ · pubmed ↗