A Case Report: Identification of a Pathogenic Microdeletion at Chromosome 21q21.3q22.13 Using Whole-Exome Sequencing and CNV Analysis in a Moroccan Child with Global Developmental Delay
Farah Jouali, Ghyzlane El Haddoumi, Imane Antra, Rachid Benhida, Afaf Ben Itto, Jamal Fekkak

TL;DR
A Moroccan child with developmental delays was found to have a pathogenic microdeletion on chromosome 21q21.3–q22.13 using whole-exome sequencing and CNV analysis.
Contribution
This case report highlights the use of integrated genomic and bioinformatic approaches to identify a rare microdeletion and its clinical implications.
Findings
An 8.2 Mb de novo microdeletion in 21q21.3–q22.13 was identified in a child with global developmental delay and multiple anomalies.
The deletion encompassed 124 clinically relevant genes, including dosage-sensitive genes like SON and RUNX1.
Integrated analysis confirmed the deletion's pathogenicity and genotype–phenotype correlations.
Abstract
Copy number variations (CNVs) affecting the chromosomal region 21q21.3–q22.13 are rare and have been increasingly associated with neurodevelopmental abnormalities and multisystemic manifestations. In this study, we aimed to characterize the clinical, genomic, and genotype–phenotype correlations of a Moroccan child carrying a de novo microdeletion in this region. Whole exome sequencing (WES) was performed using sequencing-by-synthesis technology on the GenoLab M platform, and CNV detection was achieved through the SeqOne platform. Variant interpretation was conducted using the Integrative Genomics Viewer (IGV), and a custom gene–phenotype heatmap was generated in R (ComplexHeatmap and pheatmap) based on OMIM, ClinVar, and DECIPHER databases to prioritize candidate genes within the deleted segment. The patient presented with global developmental delay, microcephaly, psychomotor and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
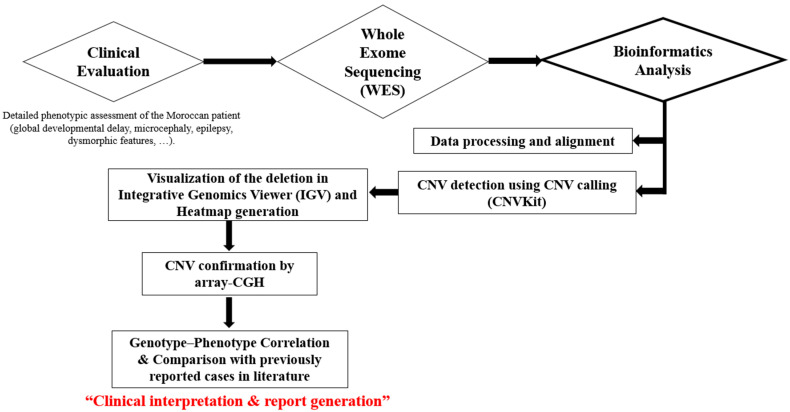 Figure 1
Figure 1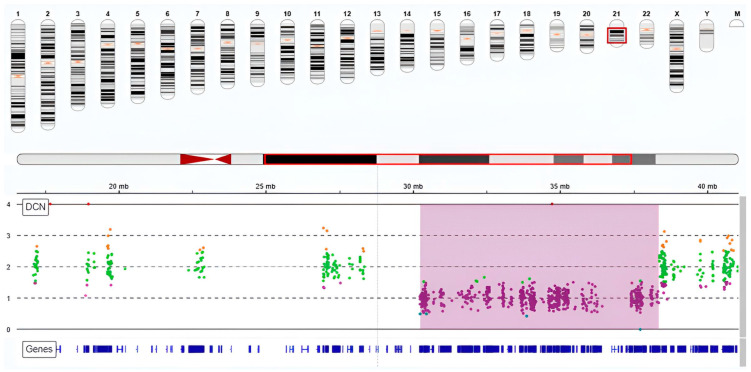 Figure 2
Figure 2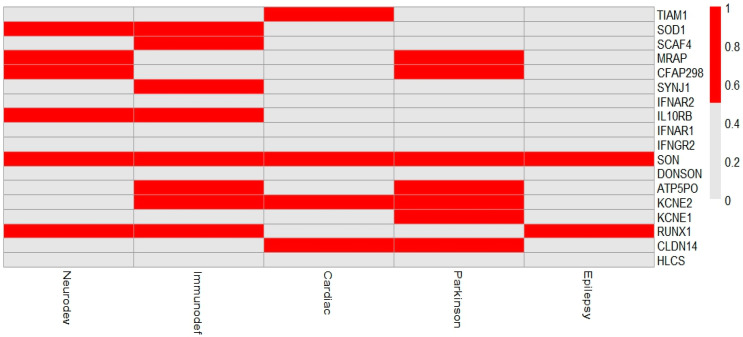 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCongenital heart defects research · Genomic variations and chromosomal abnormalities · Genomics and Rare Diseases
1. Introduction
Microdeletions affecting the chromosome 21q21.3 to 21q22.13 are infrequent but carry notable clinical implications [1]. These deletions have been associated with complex neurodevelopmental phenotypes, most commonly global developmental delay (GDD). GDD is a heterogeneous clinical condition characterized by significant delays in multiple developmental domains, including motor, speech, cognition, and social functioning. It affects approximately 1–3% of children under the age of five and often reflects an underlying genetic etiology, particularly when associated with dysmorphic features, microcephaly, or neurological manifestations such as epilepsy [2].
In this study, we identified a copy number variant (CNV) consistent with the genetic diagnosis of 21q22.11q22.12 microdeletion syndrome (ORPHA: 261323). This CNV encompasses several genes, including RUNX1 and SON. Additionally, the CNV involves genes with LOC or open reading frame (ORF) designations, including LOC101928107, Chromosome 21 open reading frame 62 (C21orf62), and others.
Among the genes located within this critical region, SON emerges as a key contributor to the aforesaid phenotypes [3]. This gene (MIM #182465), located at 21q22.11, comprises 12 regular exons, among which exon 3 is the largest one, accounting for 82% of the coding region [4,5]. It encodes a DNA- and RNA-binding protein that functions as a splicing cofactor, playing a crucial role in RNA splicing, neurodevelopment, and cell cycle regulation [6]. The canonical isoform encoded by NM_138927.2 is a protein of 2426 amino acids that is highly conserved and ubiquitously expressed across all tissues and brain cells. This protein is a central component of nuclear speckles with SRRM2 (Serine/Arginine Repetitive Matrix 2) [7,8], and controls RNA splicing, gene transcription, and stem cell maintenance [9]. Deletion of SON results in double-strand DNA breakage, disrupted microtubule dynamics, neural abnormalities, and altered cell morphology [8,9,10]. Together, these effects support its pivotal role in preserving proper cell activity and genome integrity.
SON-related syndrome, also known as Zhu–Tokita–Takenouchi–Kim (ZTTK) syndrome (MIM #617140), is a multisystemic disorder that affects less than one in a million individuals worldwide [11]. Clinical manifestations are variable but commonly include developmental delay, intellectual disability, hypotonia, seizures, and brain malformations [11]. Additional features may encompass dysmorphic craniofacial traits, ocular problems, musculoskeletal anomalies, short stature, and congenital heart and genitourinary system defects [5,12]. Zhu et al. were the first to report this autosomal dominant hereditary disease by revealing de novo truncating mutations [13,14], and a dedicated website, ZTTK SON-Shine Foundation, is available at https://zttk.org/ (accessed on 2 August 2025).
Advancements in next-generation sequencing (NGS) technologies, especially whole-exome sequencing (WES), have significantly improved the detection of undiagnosed neurodevelopmental disorders. In particular, WES combined with CNV analysis allows the detection of both single-nucleotide variants (SNVs) and larger genomic rearrangements that may underlie complex phenotypes [15,16].
To the best of our knowledge, this is the first reported case of a Moroccan child carrying a novel 8.2 Mb microdeletion at chromosome 21q21.3–q22.13, encompassing 124 genes and consistent with a rare 21q microdeletion syndrome. Although deletions within chromosome 21q have been previously associated with neurodevelopmental phenotypes, the critical regions and their full clinical spectrum remain poorly defined. In this study, we combined whole-exome sequencing (WES) with copy-number-variation (CNV) calling to investigate the potential impact of this deletion on the patient’s clinical presentation and to refine the genotype–phenotype correlation. The integration of CNV analysis with phenotypic data and gene prioritization highlights the diagnostic value of WES-based CNV detection, even in the absence of detectable single-nucleotide variants. Moreover, this work provides new insights into the contribution of dosage-sensitive genes, particularly SON and RUNX1, to the observed phenotype, thereby expanding the molecular and clinical understanding of 21q21.3–q22.13 deletions.
2. Materials and Methods
2.1. Clinical Features of the Patient
The patient, a two-year-old Moroccan child born to a non-consanguineous marriage, is the youngest of four siblings from the same parents. Clinically, the patient displayed global psychomotor and stature-ponderal retardation, alongside microcephaly, facial dysmorphia, and pharmaco-sensitive epilepsy. Imaging via brain MRI (magnetic resonance imaging) identified passive biventricular hydrocephalus and diffuse cortical atrophy.
2.2. Genomic Analysis
2.2.1. DNA Extraction
Genomic DNA was extracted from peripheral blood using a solid-phase extraction (SPE) method (Wizard^®^ Genomic DNA Purification Kit, Promega Corporation, Madison, WI, USA), following the manufacturer’s protocol.
2.2.2. Whole Exome Sequencing (WES) and Bioinformatic Analysis
The libraries were prepared on the Magnis NGS prep system using the SureSelect XT HS2 DNA Target Enrichment V8 kit with a version B0 protocol, May 2023 (Agilent Technologies, Santa Clara, CA, USA), and then quantified using the Qubit HS (Thermo Fisher Scientific, Waltham, MA, USA) and the Agilent D2400 bio-Analyzer.
Whole exome sequencing was performed using the Genolab M platform, generating 150 bp paired-end reads (2 × 150 bp), in accordance with the manufacturer’s protocol (Genemind Biosciences Co., Shenzhen, China). The raw sequencing data (FASTQ files) were uploaded to the SeqOne platform to provide a whole bioinformatic analysis, including read alignment to the human reference genome (GRCh37/hg19), variant calling, and annotation (gene, functional effect, and clinical impact). Candidate variants were then classified according to the American College of Medical Genetics and Genomics (ACMG) criteria.
2.2.3. CNV Detection and Visualization
CNV analysis was performed to detect and identify potential structural variants, thereby annotating large copy number alterations. Manual confirmation and visualization of this deletion were performed using Integrative Genomics Viewer (IGV).
2.2.4. SNP-CGH Array
A SNP-CGH array was performed to confirm the presence of this CNV at chromosome cytobands 21q21.3–q22.13. The Infinium Global Diversity Array with Cytogenetics kit (Illumina Inc., San Diego, CA, USA) was used. Genomic DNA was amplified, fragmented, and hybridized onto the array according to the manufacturer’s instructions. Data analysis was carried out using the NxClinical software 6.0 (BioDiscovery, Panchkula, India).
2.2.5. Gene–Phenotype Prioritization and Heatmap Generation
Genes encompassed within the deleted region were extracted based on genomic coordinates. Their known clinical associations were curated through manual review of OMIM, DECIPHER, ClinVar, and relevant publications indexed in PubMed. Each gene was then scored across five phenotypic categories: Neurodevelopmental disorders, Immunodeficiency, Cardiac anomalies, Epilepsy, and Parkinsonism using a binary matrix (1 = known association, 0 = no established link). The outputs were visualized as a heatmap using the ComplexHeatmap and pheatmap packages in R (v4.3.0).
2.3. Workflow Overview
To provide a clear overview of the analytical strategy, we summarized the methodological pipeline in Figure 1. The workflow includes the main steps from clinical assessment to genomic data analysis, CNV confirmation, and genotype–phenotype correlation.
3. Results
Whole-exome sequencing (WES) analysis did not identify any pathogenic single-nucleotide variants (SNVs) that could explain the patient’s phenotype. However, CNV calling based on WES read-depth data revealed a heterozygous microdeletion on the long arm of chromosome 21, specifically spanning the 21q21.3–q22.13 region (Figure 2). This deletion measures approximately 8.2 Mb, extending from position 30.3 Mb to 38.5 Mb (Table 1), and affects several clinically relevant genes, including SON and RUNX1, and has a sample frequency of 0.81%. CNV calling thresholds and quality metrics were applied using SeqOne, and visualization and confirmation were performed with IGV (Table 2).
Phenotype–Genotype Correlation
Figure 3 illustrates the distribution of phenotype associations for each gene in the deleted region.
To contextualize this result, we compared the present 8.2 Mb 21q21.3–q22.13 deletion (arr[GRCh37]21q21.3q22.13(30093156_38340656)x1) with previously reported 21q deletions and SON-related cases. Overlapping large interstitial deletions involving 21q22 that include dosage-sensitive genes such as RUNX1 have been associated with neurodevelopmental delay, dysmorphism, and hematologic abnormalities, and contiguous-gene 21q22 deletions of comparable size (~7–8 Mb) have been reported previously. Conversely, most SON-related reports describe single-gene loss-of-function (frameshift/nonsense) rather than large deletions; these studies nevertheless support the link between SON haploinsufficiency and neurodevelopmental features (developmental delay, intellectual disability, hypotonia, seizures). We therefore present a comparative table (Table 3) summarizing our case and representative previously published cases to clarify overlapping features and differences
4. Discussion
Over the past decade, the study of structural variation in the human genome has emerged as one of the fastest-growing fields in genetics. Among these variations, copy number variants (CNVs) occur more frequently than previously anticipated. While CNV contributes to interindividual phenotypic variability, they are increasingly recognized for their role in modulating the expressivity and severity of chromosomal disorders. Consequently, understanding the implications of CNV in human disease requires a careful assessment of its potential phenotypic effects. Notably, CNVs involving deletions or duplications of dosage-sensitive genes, or their regulatory regions, can disrupt gene expression or function, often resulting in significant clinical consequences [22,23].
A compelling example of the clinical relevance of CNV centers on the 21q22.11q22.12 microdeletion, a rare genetic abnormality with a distinct phenotypic profile. This syndrome results from a partial deletion of the long arm of chromosome 21 and is characterized by prenatal and postnatal growth retardation, short stature, intellectual disability, developmental delay with severe language impairment, thrombocytopenia, and craniofacial dysmorphism that may include microcephaly, downward slanting palpebral fissures, broad or depressed nasal bridge, and epicanthal folds [24]. Brain abnormalities on MRI (such as agenesis of the corpus callosum), behavioral disturbances, and seizures may be associated [24,25]. Katzaki et al. reported three new patients with overlapping de novo interstitial deletions involving cytoband 21q22 and including the RUNX1 gene, presenting with severe developmental delay, dysmorphic features, behavioral problems, and thrombocytopenia [26,27,28,29].
According to the current literature and genetic databases, the CNV identified in this case has not been previously reported and is classified as pathogenic according to ACMG guidelines. This deletion includes both the RUNX1 and SON genes, which are functionally interacting and contribute to the patient’s combined neurodevelopmental and hematologic phenotype. Both SON and RUNX1 have established haploinsufficiency evidence according to the ClinGen dosage sensitivity map. Haploinsufficiency of RUNX1 leads to reduced transcriptional regulation of key genes involved in hematopoiesis, resulting in hereditary thrombocytopenia, characterized by a decreased number of circulating platelets. Furthermore, impaired RUNX1 function disrupts the normal differentiation and proliferation of hematopoietic stem and progenitor cells, thereby increasing the risk of developing hematologic malignancies, particularly myeloid leukemias [30,31,32], while SON is implicated in Zhu–Tokita–Takenouchi–Kim syndrome [11], which is a rare neurodevelopmental disorder characterized by a heterogeneous spectrum of phenotypic and clinical features [4,6]. Additionally, SON emerged as a key node across multiple phenotypic domains, as shown in the generated heatmap, highlighting its central role in neurodevelopmental, epileptic, and immune-related pathways, which supports its contribution to the patient’s clinical presentation (Figure 3).
The presence of both SON and RUNX1 within the deleted segment provides a plausible genetic basis for the proband’s combined neurodevelopmental and hematologic features. Previous studies of 21q deletions, including RUNX1, describe thrombocytopenia and predisposition to myeloid malignancies, while separate reports of SON loss-of-function variants reproduce the core ZTTK neurodevelopmental phenotype, together supporting the pathogenicity of the present contiguous deletion [18]. A direct comparison of our case with previously published key cases is provided in Table 3, highlighting similarities and differences in clinical features, genetic findings, and outcomes. This comparison facilitates the contextualization of our findings within the existing literature and underscores the unique aspects of the present case.
Overall, the integration of molecular findings with clinical observations is crucial for refining the diagnosis of rare disorders like ZTTK and microdeletion syndromes. Although these conditions share overlapping features, such as global developmental delay, intellectual disability, hypotonia, and dysmorphic facial features, they are genetically distinct. While mutations in the SON gene have been recognized as causative of ZTTK [14], the 21q21.3–q22.13 microdeletion syndrome arises from a larger chromosomal deletion implying multiple genes leading to a more complex and heterogeneous picture [1].
In this report, our patient fits the known clinical presentation of microdeletion syndrome and highlights the broader gene loss seen in 21q microdeletions. To the best of our knowledge, this is the first case to be reported in Morocco, broadening the geographical and ethnic spectrum. The collected data emphasize the importance of establishing genotype-phenotype correlations and shed light on the critical role of genetic testing in diagnosing rare and atypical neurodevelopmental disorders across diverse populations. These findings support the pathogenicity of the identified CNV and its contribution to the neurodevelopmental and hematologic abnormalities observed.
5. Conclusions
Numerous regions of the human genome, comprising hundreds of genes, exhibit copy-number variations through recurrent deletions or duplications, highlighting their clinical relevance in shaping phenotypic variability in aneuploidy and other chromosomal disorders. In this case report, we identified a de novo heterozygous CNV in the 21q21.3–q22.13 region, encompassing key genes including SON and RUNX1, which are functionally relevant to neurodevelopmental and hematopoietic pathways. The proband’s clinical presentation illustrates the complexity and heterogeneity of microdeletion syndromes, reinforcing the syndromic nature of this CNV. Integration of whole-exome sequencing with CNV analysis, functional annotation, and gene–phenotype heatmaps further supports the pathogenicity of the deletion and its contribution to the combined neurodevelopmental and hematologic phenotype. Given that the clinical features can vary depending on the size of the deletion and the specific genes involved, close clinical follow-up is essential for monitoring potential comorbidities. These findings contribute to the growing literature on gene haploinsufficiency as a driver of multisystemic phenotypes and emphasize the need for ongoing variant curation to refine our understanding of genotype-phenotype correlations. Future functional studies will be crucial to support targeted therapies and personalized care strategies. In this context, genetic counseling remains integral, guiding reproductive choices and long-term clinical management, particularly in regions with limited access to advanced genomic technologies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Carvill G.L. Mefford H.C. Microdeletion Syndromes Curr. Opin. Genet. Dev.20132323223910.1016/j.gde.2013.03.00423664828 · doi ↗ · pubmed ↗
- 2Modha B. Global Developmental Delay and Its Considerations in Paediatric Dental Care—A Case Report Oral 2021118118910.3390/oral 1030018 · doi ↗
- 3Tokita M.J. Braxton A.A. Shao Y. Lewis A.M. Vincent M. Küry S. Besnard T. Isidor B. Latypova X. Bézieau S. De Novo Truncating Variants in SON Cause Intellectual Disability, Congenital Malformations, and Failure to Thrive Am. J. Hum. Genet.20169972072710.1016/j.ajhg.2016.06.03527545676 PMC 5011061 · doi ↗ · pubmed ↗
- 4Dingemans A.J.M. Truijen K.M.G. Kim J.-H. Alaçam Z. Faivre L. Collins K.M. Gerkes E.H. van Haelst M. van de Laar I.M.B.H. Lindstrom K. Establishing the Phenotypic Spectrum of ZTTK Syndrome by Analysis of 52 Individuals with Variants in SON Eur. J. Hum. Genet.20223027128110.1038/s 41431-021-00960-434521999 PMC 8904542 · doi ↗ · pubmed ↗
- 5Kim J.-H. Shinde D.N. Reijnders M.R.F. Hauser N.S. Belmonte R.L. Wilson G.R. Bosch D.G.M. Bubulya P.A. Shashi V. Petrovski S. De Novo Mutations in SON Disrupt RNA Splicing of Genes Essential for Brain Development and Metabolism, Causing an Intellectual-Disability Syndrome Am. J. Hum. Genet.20169971171910.1016/j.ajhg.2016.06.02927545680 PMC 5011044 · doi ↗ · pubmed ↗
- 6Slezak R. Smigiel R. Rydzanicz M. Pollak A. Kosinska J. Stawinski P. Malgorzata Sasiadek M. Ploski R. Phenotypic Expansion in Zhu-Tokita-Takenouchi-Kim Syndrome Caused by de Novo Variants in the SON Gene Mol. Genet. Genomic. Med.20208 e 143210.1002/mgg 3.143232705777 PMC 7549597 · doi ↗ · pubmed ↗
- 7Ilikİ.A. Malszycki M. Lübke A.K. Schade C. Meierhofer D. AktaşT. SON and SRRM 2 Are Essential for Nuclear Speckle Formatione Life 20209 e 6057910.7554/e Life.6057933095160 PMC 7671692 · doi ↗ · pubmed ↗
- 8Xu S. Lai S.-K. Sim D.Y. Ang W.S.L. Li H.Y. Roca X. SRRM 2 Organizes Splicing Condensates to Regulate Alternative Splicing Nucleic Acids Res.2022508599861410.1093/nar/gkac 66935929045 PMC 9410892 · doi ↗ · pubmed ↗