Genetic Insights into Familial Hypospadias Identifying Rare Variants and Their Potential Role in Urethral Development
Kholoud N. Al-Shafai, Seem Arar, Asma Jamil, Amina Azzah, Maraeh Mancha, Luis R. Saraiva, Tariq Abbas

TL;DR
This study identifies rare genetic variants linked to hypospadias, a birth defect in males, offering new insights into its genetic causes and potential for future treatments.
Contribution
The study reports novel likely pathogenic variants in genes (EIF2B5, INO80, ACADVL) not previously associated with hypospadias.
Findings
Three likely pathogenic variants in EIF2B5, INO80, and ACADVL genes were identified in index patients and co-segregated with hypospadias.
Variants of uncertain significance were found in DNAH12, LHFP, and COL6A3, which may contribute to the hypospadias phenotype.
Two families showed no potential causative variants, highlighting the need for further analysis of copy number variants.
Abstract
Background: Hypospadias is a common congenital condition in male infants, characterised by incomplete development of the underside of the penile shaft. Genetic factors play a major role in its development. Therefore, studying genetic contributions, especially in familial cases, can enhance our understanding of disease causes and guide targeted interventions. Materials and Methods: Through a structured biobank for hypospadias, we collected blood samples from individuals with familial hypospadias and their relatives. Whole-genome sequencing (WGS) was performed on 27 individuals across seven families to identify potential genetic causes. Bioinformatics analysis, including the GEMINI tool, was used to assess inheritance patterns of single-nucleotide variants (SNVs) within families and identify potential causative SNVs. Results: We identified three likely pathogenic variants in genes not…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
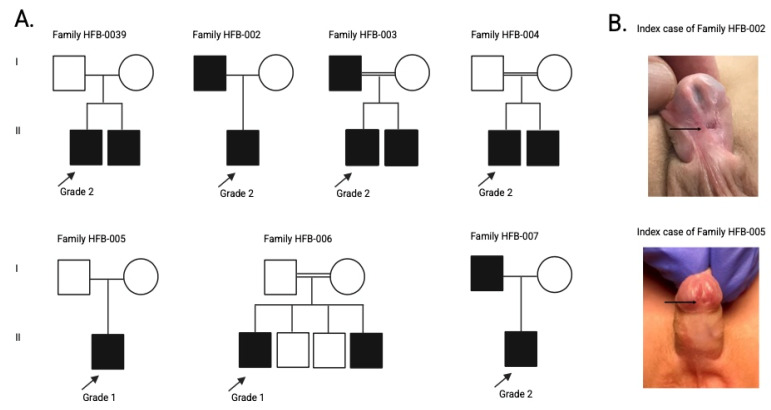 Figure 1
Figure 1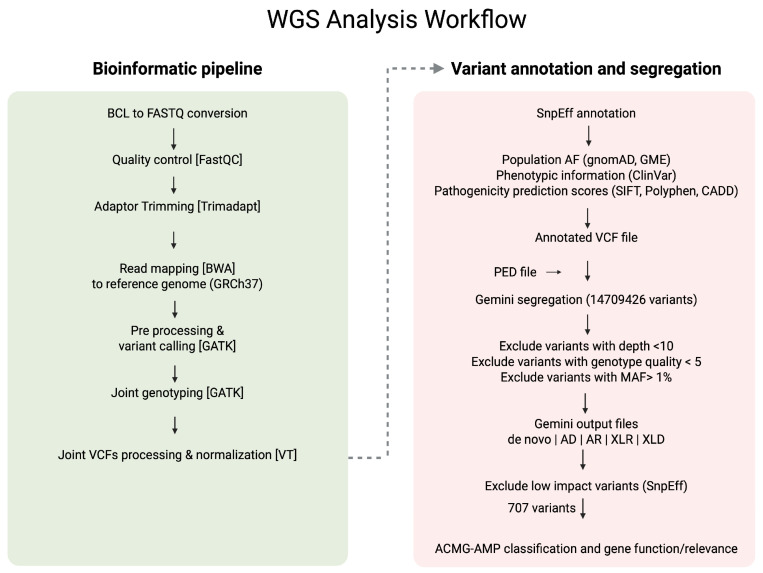 Figure 2
Figure 2- —Sidra Medicine, Doha, Qatar
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsUrological Disorders and Treatments · Tissue Engineering and Regenerative Medicine · Pediatric Urology and Nephrology Studies
1. Introduction
Hypospadias is among the most common congenital anomalies in males, occurring in about 2 out of every 1000 pregnancies, including live births, stillbirths, and terminations [1]. It is characterised by the ectopic placement of the urethral meatus on the ventral side of the penis, often accompanied by chordee and an abnormal foreskin. Despite surgical correction in early childhood, many individuals face long-term complications affecting urinary, reproductive, and sexual health [2]. Clinical management depends on accurate diagnosis and classification based on meatal location and severity, which guides the choice of surgical technique and helps predict outcomes [3].
While environmental and endocrine-disrupting exposures have been implicated, familial clustering and twin studies strongly support a genetic contribution to hypospadias, with heritability estimates ranging between 57% and 77% [4,5,6]. The development of male external genitalia critically depends on androgen signalling, particularly the actions of testosterone and its more potent derivative, dihydrotestosterone (DHT). Disruptions in this signalling axis caused by pathogenic variants in genes such as Androgen Receptor (AR) and Steroidogenic Factor 1 (SF1/NR5A1) can lead to incomplete virilisation and structural anomalies [7,8]. To date, at least 49 genes have been linked to hypospadias pathogenesis, covering androgen metabolism, hormone receptor activity, and urogenital development [9]. Furthermore, genome-wide association studies (GWASs) in sporadic hypospadias have identified several susceptibility loci containing developmental regulators including HOXA4, IRX5, and EYA1 [4]. However, the genetic basis of hypospadias remains only partially understood, particularly in non-European populations.
To address this gap, we recently established the Hypospadias Biobank Cohort (HBC), which is a structured, longitudinal research platform integrating detailed clinical, phenotypic, and surgical data with biological specimens [10]. The HBC enables the systematic investigation of genotype–phenotype correlations and supports the refinement of classification frameworks for hypospadias severity and outcomes. As part of this effort, we recruited multiplex families with familial hypospadias and collected biospecimens from affected individuals and their relatives. In this study, we report whole genome sequencing (WGS) data analysis from 27 individuals across seven families within the HBC, aiming to identify rare or high-impact genetic variants that may underlie hypospadias pathogenesis in this understudied population, where inbreeding and consanguinity are common practices that would influence the genetic architecture of hypospadias, as noticed for other disease phenotypes [11].
2. Material and Methods
2.1. Ethical Approval
Ethical approval was obtained from the Institutional Review Board of Sidra Medicine (IRB #1866246), and the study was conducted in accordance with the Declaration of Helsinki. Informed consent/assent was obtained from all study participants to establish a structured Hypospadias Biobank Cohort (HBC) at Sidra Medicine in Qatar, for which comprehensive data and samples were collected from participants for research purposes, as detailed elsewhere [10].
Participants in the HBC were selected based on specified inclusion criteria to ensure broad representation across the spectrum of hypospadias, including distal, midshaft, and proximal forms [10]. Urethral defect ratio (UDR) was calculated by dividing the extent of the urethral defect (distance between the glandular knobs and BCS) by the stretched penile length (SPL). Hypospadias severity was then classified into three distinct grades (UDR < 0.5, 0.5–0.99, ≥1.0). Participants with additional syndromic conditions were excluded to reduce the complexity of genetic analyses, enabling a more focused examination of isolated hypospadias. Finally, participants from familial cases of hypospadias, where more than one family member has hypospadias, were considered eligible, as this suggests a potential genetic predisposition. Individuals with syndromic conditions or known genetic disorders that could interfere with future analyses were excluded, along with eligible participants who were unable to provide informed consent or assent due to language barriers or medical reasons.
2.2. Study Participants
Seven familial cases of hypospadias and their relatives (total n = 27) were identified and enrolled at HBC for sample and data collection, followed by genetic investigation. Familial cases of hypospadias refer to situations in which more than one family member is affected by the same condition, suggesting a genetic or hereditary basis. The 27 study participants included 7 index cases and 20 of their family members (7 affected, 13 unaffected). Family pedigrees are provided in Figure 1.
2.3. Whole Genome Sequencing (WGS) Data Generation and Processing
A total of 4–10 mL of peripheral blood samples, collected into EDTA-containing tubes, were obtained from each participant for genomic DNA extraction, followed by WGS library preparation using the Truseq DNA Nano Kit (Illumina, San Diego, CA, USA) at the Sidra Integrative Services Laboratory. Sequencing was conducted on the NovaSeq 6000 platform (Illumina), achieving an average coverage of 30–40×. Our analysis pipeline involved demultiplexing the raw sequencing data (.bcl files) into FastQ files. Read quality was evaluated with FastQC (https://github.com/s-andrews/FastQC, accessed on 1 July 2024), and adapter sequences were trimmed using Trimadap (https://github.com/lh3/trimadap, accessed on 1 July 2024). High-quality reads were aligned to the human reference genome GRCh37/hg19 (NIH, New York, NY, USA) with BWA-MEM from Burrows-Wheeler Aligner v7.0.8 (Cambridge, MA, USA; arXiv:1303.3997 [q-bio.GN]). Duplicate reads were marked with SAMBLASTER v0.1.22, and alignment files were sorted using SAMtools from the BWA-kit package [12]. Picard and Mosdepth were utilised for additional quality control of the alignments. After base quality score recalibration, variants were called with GATK v4.0.9.0 [13]. Individual gVCF files were jointly called to produce a merged VCF file. These joint VCFs were split, decomposed, and normalised using Variant Transformer v0.57 [14]. Variants were annotated with SnpEff v4.3 [15], allele frequency databases such as gnomwAD [16] and the Greater Middle Eastern [17], clinical databases like ClinVar [18], and pathogenicity prediction scores including SIFT [19], PolyPhen-2 [20], and CADD [21].
2.4. Single-Nucleotide Variant (SNV) Segregation Analysis and Filtration
Segregation analysis of single nucleotide variants (SNVSs) was performed using Gemini v0.30.2 [22]. The annotated VCF files, along with corresponding pedigree information, were imported into the Gemini platform using the vcf2db utility. This facilitated the exploration of various inheritance models, including de novo, autosomal dominant, autosomal recessive, compound heterozygous, X-linked recessive, and X-linked dominant patterns. A custom Python v.3.9.0 script was employed to export Gemini query results into Excel spreadsheets for further filtering and interpretation. Each family was analysed based on the potential mode of inheritance suggested by the pedigree information. Variants were excluded if they had a read depth less than10, genotype quality less than 5, a minor allele frequency (MAF) above 1% in population databases, or were predicted to have low functional impact according to SnpEff annotations. The remaining variants were subsequently evaluated and classified following the ACMG-AMP guidelines [23] using Varsome [24] and Franklin (https://franklin.genoox.com/clinical-db/home, accessed on 1 July 2024), with a literature review and gene function assessment. Figure 2 provides a clear illustration of the variant filtering and analysis workflow that was used in the study.
3. Results
3.1. Demographic and Clinical Characteristics
For this study, a total of 27 individuals were enrolled across the seven families, comprising the seven index cases and 20 relatives (7 affected, 13 unaffected), to enable combined clinical and genetic analyses (Figure 1). The demographic and clinical features of the seven index cases with familial hypospadias enrolled in this study are summarised in Table 1. The median age at diagnosis was 3 years (range: 2–10 years). All index cases were male, and the cohort encompassed a diverse set of ethnic backgrounds, including Sudanese, Indian, Pakistani, Palestinian, Yemeni, and Egyptian nationalities. Five of the seven probands (71%) were of Arab ethnicity, reflecting the population structure. Parental consanguinity was reported in three families, consistent with the high consanguinity rates observed in the Middle Eastern countries [25] and Pakistan [26]. All index cases had a positive family history of hypospadias, supporting a likely genetic aetiology. Treatment included surgical correction in all cases, with two patients also receiving preoperative intramuscular testosterone injections of 2 mg/kg twice monthly.
3.2. Variants Detected from WGS Analysis
WGS of the seven families with familial hypospadias described above identified multiple rare variants, some with potential pathogenic relevance (Figure 2 and Table 2). Supplementary Figure S1 provides the Integrated Genomics Viewer (IGV) screenshots of the described variants, in the index cases and relatives. Two of these variants were classified as pathogenic (P) and three as likely pathogenic (LP), along with four variants of uncertain significance (VUS), some of which may be pathogenic and are located in genes not previously linked to hypospadias aetiology.
3.3. Pathogenic and Likely Pathogenic Gene Variants
Three out of the seven families had at least one pathogenic or likely pathogenic variant identified. In Family HFB-002, a pathogenic splice site gene variant (c.642+1G>A) and a likely pathogenic missense variant (c.821G>A) were identified in TTC37 and EIF2B5 genes, respectively. These variants were inherited from the affected father in a heterozygous state. The TTC37-encoded protein is involved in RNA degradation activities [27]. Mutations in the TTC37 gene have been linked to Trichohepatoenteric syndrome (syndromic diarrhoea) in both homozygosity and compound heterozygosity [28]. The variant has a high SpliceAI score (0.94), suggesting it would influence splicing activity [29]. The protein encoded by EIF2B5 is critical for recycling GDP-bound eIF2 into its active GTP-bound form, an essential step in initiating protein synthesis. Mutations in EIF2B5 impair the regulation of protein synthesis during cellular stress, leading to vanishing white matter disease, a progressive leukodystrophy characterised by neurological deterioration [30]. A VUS was also detected in this family within the DNAH12 gene, as described later in this manuscript.
In Family HFB-003, a heterozygous frameshift variant (c.458dupG) in the OBSL1 gene, classified as pathogenic, was identified. OBSL1 encodes obscurin-like protein 1, a cytoskeletal protein involved in muscle structure and function. A recent study demonstrated that a homozygous OBSL1 mutation (c.848delG) can cause Three M Syndrome 2 (3M2), a rare growth disorder characterised by short stature and distinctive facial and skeletal features, in a Pakistani family. Interestingly, affected members of that family also exhibited atypical features, including hypospadias [31]. Furthermore, a heterozygous missense variant (c.445G>C) in the INO80 gene, classified as likely pathogenic, was also detected in Family HFB-003. INO80 encodes a chromatin remodelling factor that regulates Bone Morphogenetic Protein 4 (BMP4) expression, a well-established gene involved in urethral development and hypospadias pathogenesis [32,33].
In Family HFB-007, a likely pathogenic heterozygous variant (c.67C>T) was identified in ACADVL, both of which co-segregated with the phenotype within the family. The ACADVL gene encodes the enzyme very long-chain acyl-CoA dehydrogenase, which is vital for the mitochondrial β-oxidation of very long-chain fatty acids. Mutations in ACADVL cause the autosomal recessive disorder very long-chain acyl-CoA dehydrogenase deficiency (VLCADD; OMIM #201475), which can manifest as early onset cardiomyopathy, hypoketotic hypoglycaemia, or adult-onset rhabdomyolysis.
3.4. Variants of Uncertain Significance (VUS)
Two homozygous VUS were identified in two of the studied families (Family HFB-0039 and Family HFB-004). In Family HFB-0039, a rare homozygous splice site variant (c.-176A>G) in the LHFP gene was identified in the two affected males of one family, while the unaffected parents were heterozygous for this variant. Although the LHFP gene itself has not been previously linked to hypospadias, functional studies of related gene family members such as LHFPL2 have reported causes of hypospadias-like phenotypes in mice and suggested a mechanistic role of the encoded protein in genital development [34]. Moreover, in Family HFB-004, a homozygous missense variant (c.3223C>T) of uncertain significance (VUS) was identified in COL6A3, a gene implicated in extracellular matrix integrity but not previously linked to genital malformations. However, COL6A3 gene expression was upregulated in DNAH17 mutants detected in severe hypospadias [7] and therefore requires further assessment.
In addition to these two homozygous variants, a heterozygous missense VUS variant (c.7604C>T) in DNAH12 was identified in Family HFB-002, in both the index case and his affected father. The DNAH12 gene encodes a dynein axonemal heavy chain, a motor protein involved in microtubule-based movement and ciliary function, including sperm motility. Although rare damaging variants in several dynein heavy chain genes have been linked to hypospadias, DNAH12 itself has not been specifically implicated, and a recent study found no enrichment of DNAH12 mutations in severe hypospadias cases compared with the controls [7]. Interestingly, a recent study reported that male mice lacking Dnah12 were infertile and displayed smaller testes with reduced sperm counts, whereas females developed normally [35].
No candidate variants were identified in two related families (Family HFB-005 and Family HFB-006), suggesting the need for further investigation into non-coding variants, regulatory elements, or structural variation such as copy number variants (CNVs). The absence of non-coding region and CNV analyses represents a limitation of this study, as regulatory or structural variants may underlie the observed phenotypes. Future functional validation experiments will be essential to confirm the pathogenicity of the prioritised variants and to establish mechanistic links to hypospadias development.
4. Discussion
The findings from this study offer important insights into the genetic foundations of familial hypospadias and its potential hereditary causes in highly inbred and understudied populations. In this cohort of seven families with familial hypospadias, we identified several rare variants with likely pathogenic significance, located in genes that have not previously been linked to the hypospadias phenotype. These results not only add to the growing body of knowledge on the genetic basis of hypospadias but also emphasise the complexity and diversity of its aetiology, potentially involving both common and rare genetic variants that influence urethral and genital development.
The INO80 missense variant (c.445G>C) also seems promising, as INO80 is involved in chromatin remodelling and regulates BMP4, a key gene in urethral development [32,33]. The role of chromatin remodelling factors in hypospadias pathogenesis has not been extensively studied, making this a potentially novel and important pathway to explore in future research. The same family also has the OBSL1 frameshift variant, with previous evidence suggesting that mutations in this gene can be associated with the hypospadias phenotype [31].
Among the identified variants, three VUS variants stood out. This includes the homozygous LHFP splice site mutation found in family HFB-0039, which is especially noteworthy. Although LHFP has not previously been linked to hypospadias, functional studies on related genes (LHFPL2) suggest a role in genital development [34]. Specifically, LHFPL2 mutant mice exhibit elongation of Müllerian and Wolffian ducts; however, their duct tips are enlarged and fail to merge with the urogenital sinus, emphasising the importance of this gene in distal reproductive tract development [34]. This finding raises the possibility that mutations in LHFP, such as the identified variant c.-176A>G, may contribute to the development of hypospadias, warranting further investigation. Additionally, the identification of another homozygous variant of uncertain significance (VUS) in COL6A3, a gene previously implicated in extracellular matrix integrity, highlights a potential new area of interest warranting further investigation. Notably, COL6A3 expression has been reported to be upregulated in DNAH17 mutants associated with severe hypospadias [7], suggesting a possible link that merits closer examination. Notably, neither the LHFP nor the COL6A3 variants were detected as homozygous in gnomAD v4.1.0 [36]. Also, a heterozygous missense VUS (c.7604C>T) in DNAH12 was identified in both the index case and his affected father in Family HFB-002. Although DNAH12 has not been directly implicated in hypospadias, its role in ciliary function and sperm motility suggests potential relevance to male reproductive development. Notably, animal studies have shown that male Dnah12-knockout mice are infertile and exhibit testicular abnormalities, supporting a possible role in testis or sperm function [35]. However, current human data do not indicate a significant enrichment of DNAH12 variants in hypospadias cases [7]. Therefore, while this variant remains of uncertain significance, its biological function warrants further investigation, particularly in the context of ureteral development.
Although these findings offer valuable insights, the identification of VUS in LHFP, COL6A3, and DNAH12 highlights the importance of caution when interpreting the clinical relevance of these genetic changes, and further validation is needed to establish their pathogenicity.
Our results underscore the necessity for ongoing research into non-coding variants, regulatory elements, and structural variations, including CNVs [37], which might contribute to the development of hypospadias, especially in the two related families (Family HFB-005 and Family HFB-006) for which no candidate variants were identified.
From a clinical perspective, identifying genetic variants linked to familial hypospadias has the potential to significantly improve our understanding of the condition’s pathogenesis, diagnosis, and management [38]. Currently, hypospadias is diagnosed through clinical examination, and treatment mainly involves surgical correction [6]. However, increasing recognition of a genetic component in hypospadias suggests that genetic testing could become an essential diagnostic tool, especially in familial cases. Identifying specific genetic mutations could not only facilitate early diagnosis but also offer valuable insights for predicting severity and guiding treatment choices. For example, finding genetic variants associated with more severe forms of hypospadias could help clinicians customise surgical approaches based on the patient’s genetic profile. Additionally, genetic insights may enable the identification of individuals at higher risk in future pregnancies, aiding prenatal counselling and testing. Genetic testing could also help distinguish isolated hypospadias from other congenital anomalies with similar phenotypes, thereby improving clinical accuracy. Furthermore, these genetic discoveries might contribute to the development of targeted therapies, particularly when surgical correction alone is inadequate or when the condition is linked to significant comorbidities. For instance, if genes such as INO80 and BMP4 are involved in the pathogenesis of hypospadias, therapeutic strategies that target chromatin remodelling or BMP4 signalling could be explored [32,33].
While the findings from this study are promising, several avenues for future research will be essential for advancing our understanding of familial hypospadias. First, larger cohort studies, including both familial and sporadic cases, are necessary to confirm the association of the identified variants with hypospadias. Since the genetic architecture of hypospadias is likely complex, involving multiple genetic factors, further research should aim to identify additional genes and variants that may contribute to its development. Future investigations should also expand the scope of genetic analysis to encompass non-coding regions of the genome and structural variations, such as CNVs [37]. These regulatory elements and large genomic rearrangements might play a role in hypospadias development and remain largely unexplored. Additionally, functional validation studies are required to verify the pathogenicity of the identified variants and understand their molecular mechanisms. Techniques like CRISPR-Cas9-based gene editing in model organisms could help explore the functional effects of these variants on genital development. An important direction for future research is to develop improved tools for genetic counselling and prenatal screening for hypospadias. As more genetic markers are identified, creating reliable and cost-effective genetic tests suitable for clinical use will be crucial. Moreover, discovering genetic risk factors can enhance predictive models for assessing the recurrence risk of hypospadias within families, aiding family planning and early intervention. Ultimately, a multidisciplinary approach involving paediatric urologists, geneticists, and functional biologists will be vital to translating these genetic findings into clinical practice. By fostering collaboration across specialities, we can ensure that insights from genetic research are effectively applied to improve care and outcomes for individuals with hypospadias.
Although the present study focused on familial cases and genetic findings, hypospadias is widely recognised as a multifactorial condition resulting from a complex interplay between genetic susceptibility and environmental exposures. Several studies have reported associations between maternal exposure to endocrine-disrupting chemicals, hormonal treatments during pregnancy, and increased risk of hypospadias [39]. Moreover, emerging evidence suggests that epigenetic mechanisms, including aberrant DNA methylation and histone modifications in developmental or hormone receptor genes, may influence urethral formation [40]. These findings underscore the potential importance of gene–environment interactions and epigenetic regulation in the aetiology of hypospadias, warranting further integrative studies to disentangle their relative contributions
5. Conclusions
This study highlights the critical role of disease biobanking and comprehensive genetic analysis in uncovering the potential underlying causes of congenital conditions such as hypospadias. By leveraging large-scale genomic data, we were able to identify specific genetic variants that may contribute to the development of this condition, offering new insights into its complex aetiology. These findings not only open avenues for future functional investigations to validate the biological relevance of these variants but also pave the way for the development of more targeted diagnostic tools and potential therapeutic strategies. Furthermore, this research underscores the broader value of integrating biobank resources with advanced genomic technologies to better understand rare and understudied congenital anomalies. Ultimately, deepening our understanding of the genetic architecture of hypospadias in understudied populations holds promise for improving early detection, personalised care, and long-term outcomes for affected individuals in this region and beyond.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Yu X. Nassar N. Mastroiacovo P. Canfield M. Groisman B. Bermejo-Sánchez E. Ritvanen A. Kiuru-Kuhlefelt S. Benavides A. Sipek A. Hypospadias Prevalence and Trends in International Birth Defect Surveillance Systems, 1980–2010 Eur. Urol.20197648249010.1016/j.eururo.2019.06.02731300237 PMC 7265200 · doi ↗ · pubmed ↗
- 2Stadler H.S. Peters C.A. Sturm R.M. Baker L.A. Best C.J.M. Bird V.Y. Geller F. Hoshizaki D.K. Knudsen T.B. Norton J.M. Meeting report on the NIDDK/AUA Workshop on Congenital Anomalies of External Genitalia: Challenges and opportunities for translational research J. Pediatr. Urol.20201679180410.1016/j.jpurol.2020.09.01233097421 PMC 7885182 · doi ↗ · pubmed ↗
- 3Abbas T. Management of Distal Hypospadias: New Insights and Stepwise Management Algorithm Hypospadiology: Current Challenges and Future Perspectives Abbas T. Springer Nature Singapore 20236780
- 4Geller F. Feenstra B. Carstensen L. Pers T.H. van Rooij I.A. Körberg I.B. Choudhry S. Karjalainen J.M. Schnack T.H. Hollegaard M.V. Genome-wide association analyses identify variants in developmental genes associated with hypospadias Nat. Genet.20144695796310.1038/ng.306325108383 · doi ↗ · pubmed ↗
- 5Schnack T.H. Zdravkovic S. Myrup C. Westergaard T. Christensen K. Wohlfahrt J. Melbye M. Familial aggregation of hypospadias: A cohort study Am. J. Epidemiol.200816725125610.1093/aje/kwm 31718042671 · doi ↗ · pubmed ↗
- 6Abbas T. Vallasciani S. Toward an Ecosystem Model of Hypospadiology Hypospadiology: Current Challenges and Future Perspectives Abbas T. Springer Nature Singapore 2023123
- 7Chen Z. Lei Y. Finnell R.H. Ding Y. Su Z. Wang Y. Xie H. Chen F. Whole-exome sequencing study of hypospadiasi Science 20232610666310.1016/j.isci.2023.10666337168556 PMC 10165268 · doi ↗ · pubmed ↗
- 8Matsushita S. Suzuki K. Murashima A. Kajioka D. Acebedo A.R. Miyagawa S. Haraguchi R. Ogino Y. Yamada G. Regulation of masculinization: Androgen signalling for external genitalia development Nat. Rev. Urol.20181535836810.1038/s 41585-018-0008-y 29670181 · doi ↗ · pubmed ↗