Otofaciocervical Syndrome and Its Overlap with Branchiootorenal Spectrum: An Integrated Literature Analysis of EYA1-Related Disorders, Including a Novel Case with an 8q13.2q13.3 Deletion
Ludovico Graziani, Miriam Lucia Carriero, Salvatore Melchionda, Bartolomeo Augello, Orazio Palumbo, Mario Bengala, Marco Castori, Giuseppe Novelli

TL;DR
This paper explores the connection between two rare genetic disorders, Otofaciocervical Syndrome and Branchiootorenal Spectrum Disorders, showing they are part of the same condition linked to the EYA1 gene.
Contribution
The study confirms that Otofaciocervical Syndrome and Branchiootorenal Spectrum Disorders are allelic disorders caused by EYA1 gene defects.
Findings
All EYA1 variant types can cause either Otofaciocervical Syndrome, Branchiootorenal Spectrum Disorders, or hybrid phenotypes.
Renal anomalies are consistently present in Otofaciocervical Syndrome patients with EYA1 variants.
The findings suggest that Otofaciocervical Syndrome and Branchiootorenal Spectrum Disorders are part of a single EYA1-related diagnostic spectrum.
Abstract
Otofaciocervical syndrome (OTFCS) is a rare disorder characterized by facial, auditory, and shoulder girdle anomalies. Its significant phenotypic overlap with branchiootorenal spectrum disorders (BORSD)—both linked to EYA1 (EYA transcriptional coactivator and phosphatase 1) gene defects—has raised questions about whether they are distinct entities or part of a single clinical spectrum. We report a novel OTFCS patient with a de novo microdeletion spanning EYA1 and review all published cases of EYA1-related disorders. Our analysis reveals that all EYA1 variant types (truncating, missense, CNV, etc.) can cause BORSD, OTFCS, or hybrid phenotypes, firmly supporting their status as allelic disorders. Crucially, all reported OTFCS patients with EYA1 variants had renal anomalies, a feature previously considered a hallmark of BORSD. We conclude that BORSD and OTFCS constitute a single…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
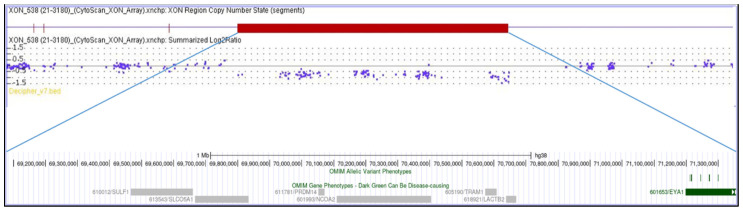 Figure 1
Figure 1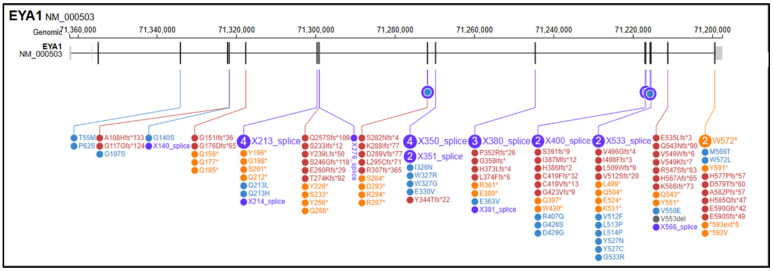 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHead and Neck Anomalies · Developmental Biology and Gene Regulation · Congenital heart defects research
1. Introduction
Craniofacial syndromes associated with branchial arch anomalies represent a clinically and genetically heterogeneous group of disorders, often characterized by overlapping features that complicate diagnosis and etiological classification [1,2]. Among these, Otofaciocervical syndrome (OTFCS) is a rare genetic disorder first described by Fara et al. in 1967, with fewer than ten cases reported in the literature [3,4]. It is characterized by peculiar craniofacial traits (e.g., long triangular face, broad forehead, narrow nose and mandible, and high arched palate), ear abnormalities (e.g., low-set, cup-shaped ears with prominent conchae and a hypoplastic tragus and lobe) often associated with hearing loss, and shoulder girdle anomalies (sloping shoulders, low-set clavicles, winged scapulae, and trapezius hypoplasia). Skeletal anomalies other than girdle anomalies and nasolacrimal duct defects are frequently reported, whereas neurodevelopmental delay and short stature are observed only in some patients [5,6]. OTFCS shares significant phenotypic overlap with branchiootorenal spectrum disorders (BORSD) [7,8]. Nonetheless, they have been previously described as clinically distinct entities: phenotypic traits such as facial dysmorphisms and shoulder girdle anomalies were considered specific to OTFCS, whereas BORSD were explicitly characterized by functional and structural renal anomalies (Table 1) [9,10].
Heterozygous variants in EYA1 (EYA transcriptional coactivator and phosphatase 1) account for approximately 40–75% of individuals clinically diagnosed with BORSD [11,12], but have also been reported in OTFCS patients [13,14,15]. Other genes in the Pax-Six-Eya-Dach network (PSEDN) are likewise implicated in both phenotypes. Heterozygous variants in SIX1 (sine oculis homeobox homolog 1) and SIX5 (sine oculis homeobox homolog 5) have been detected in 3.0–45% and 0–3.1% of individuals with BORSD, respectively [16,17,18]. In addition, biallelic PAX1 (paired box 1) variants underlie OTFCS type 2 with T-cell deficiency (OTFCS2) [19,20,21,22], while loss-of-function (LoF) variants in EYA4 (EYA transcriptional coactivator and phosphatase 4) have more recently been reported in a single affected family [4].
Whether OTFCS and BORSD represent distinct nosological entities or instead form part of a broader phenotypic continuum remains unresolved, as the precise genetic basis of OTFCS is not yet fully clarified. Importantly, some individuals with a BORSD diagnosis present with features typical of OTFCS—musculoskeletal and neurodevelopmental involvement—while some OTFCS patients exhibit renal anomalies, suggesting that the two conditions may, at least in part, represent allelic disorders [11,23,24]. This growing body of evidence supports the hypothesis that these disorders may, at least in part, represent allelic conditions.
In this study, we report a novel patient presenting with OTFCS harboring a de novo microdeletion encompassing EYA1 and perform a comprehensive review of all published cases of EYA1-related disorders. By delineating overlaps and distinctions between OTFCS and BORSD, we aim to refine their allelic relationship, improve diagnostic precision, and inform genetic counseling, while contributing to a deeper understanding of the molecular mechanisms underlying these syndromes.
2. Materials and Methods
2.1. Clinical and Molecular Data
Clinical and audiological information was collected for the index patient, including detailed phenotypic characterization with particular attention to branchial, auricular, renal, and neurodevelopmental features. Audiological assessments included the type and degree of hearing loss. Genomic DNA was extracted from peripheral blood samples.
Chromosome microarray analysis (CMA) was performed using the CytoScan XON array (Thermo Fisher Scientific, Waltham, MA, USA).
Multiplex Ligation-dependent Probe Amplification (MLPA) was performed using the SALSA MLPA probemix P153-B1 EYA1 kit (MRC-Holland, Amsterdam, The Netherlands), and variant analysis was carried out with Coffalyser.Net software v.240129.1959 (MRC-Holland, Amterdam, The Netherlands). The coordinates of detected deletions were mapped to the human genome assembly hg38 (GRCh38). Segregation analysis was performed to determine the inheritance pattern.
2.2. Literature Review and Data Extraction
A systematic literature review was conducted (last search: August 2025) using PubMed, Scopus, Embase, and Google Scholar, with the following keywords: “BORSD”, “BOR syndrome”, “BO syndrome”, “OFC syndrome”, “OTFC syndrome”, “BOF syndrome”, “BOU syndrome”, “branchio-oto-renal”, “branchio-otic”, “Otofaciocervical”, “del”, “deletion”, and “EYA1”. Filters applied: English language, human studies, and original clinical/genetic data.
2.3. Inclusion and Exclusion Criteria
Included: case reports, series, or cohorts with (i) EYA1 SNVs/indels or CNVs and (ii) patient-level clinical data covering ≥2 domains (branchial, otologic, renal, craniofacial, musculoskeletal). Excluded: reviews, functional-only/animal studies, or overlapping cohorts (retaining the most complete report).
2.4. Screening and Data Extraction
Two reviewers independently screened titles/abstracts, followed by a full-text review. Extracted data included demographics, clinical features (categorized as BORSD-typical or OTFCS-typical), variant type (missense, truncating, splice, indel, stop-loss, structural/CNV), and deletion coordinates. All variants were described according to HGVS nomenclature using the EYA1 transcript NM_000503.6 and mapped to GRCh38. Duplicates were removed.
3. Clinical Presentation
A 22-year-old female, born at term, second child of healthy non-consanguineous parents, presented with severe congenital bilateral mixed hearing loss, bilateral preauricular fistulas, hypoplasia of the left shoulder muscles, winged scapula, short stature (<3rd percentile), and a history of speech delay. Chromosome analysis revealed a normal female karyotype (46,XX). Analysis of the EYA1 gene was negative for variants using sequencing approaches. MLPA analysis identified a heterozygous de novo deletion encompassing the entire coding region of EYA1 at 8q13.3. CMA (Figure 1) confirmed a 2.3 Mb interstitial deletion at 8q13.2q13.3 chromosomal region, which spanned from nucleotides 69,068,130 to 71,362,732 (GRCh38) and involved 12 genes (LINC01592, LINC01603, SULF1, SLCO5A1, PRDM14, NCOA2, LOC101926892, TRAM1, LACTB2-AS1, LACTB2, XKR9, EYA1), and which are further characterized in Table 2. The microdeletion occurred de novo because both parents were wild-type.
4. Results
The search retrieved more than 200 records in PubMed and additional records in Scopus, Embase and Google Scholar; after deduplication and eligibility screening, 55 studies and 141 reported SNVs were included, as described in Supplementary Materials. Among these, 54 (38.3%) were frameshift variants (fs), 30 (21.3%) were nonsense variants (ns), 28 (19.9%) were splice-site variants (sp), 26 (18.4%) were missense variants (ms), 2 (1.4%) were stop-loss/stop-like variants (sl), and 1 (0.7%) was annotated as an indel (Figure 2). EYA1 gene SNVs found in the literature in association with OTFCS/BORSD spectrum are shown in Table 3, according to the first accession of genotype and/or complete phenotype. 1 splice site variant was associated with a single OTFCS case, 1 missense variant was associated with unrelated OTFCS and BORSD cases, while all the remainder SNVs were detected within the BORSD spectrum. Renal involvement is apparently absent in association with 34/141 (24.1%) described SNVs, regardless of the type of variant (ns, fs, sp, ms, sl) within the BORSD spectrum. OTFCS cases (both due to SNVs and CNVs) are further characterized in Table 4 and compared to the reported case.
In addition to the distribution of variant classes, our analysis revealed that large EYA1 deletions are enriched among BORSD cases, accounting for approximately 20% of the reports in the literature. Moreover, two-thirds of reported EYA1 SNVs were predicted to be LoF, consistent with haploinsufficiency as the main disease mechanism.
5. Discussion
The present review highlights the complex relationship between BORSD and OTFCS, both associated with EYA1 copy number and sequence variants. BORSD has traditionally been defined by a triad of branchial, otologic, and renal anomalies [7,18]. In contrast, OTFCS has been described as a distinct condition, characterized by musculoskeletal anomalies such as scapular dysplasia and short stature [9,14]. However, our systematic analysis and the present case emphasize that considerable phenotypic overlap exists, and that classical BORSD features may co-occur with OTFCS hallmarks.
The EYA proteins are components of a conserved regulatory network that is often referred to as the PAX–SIX–EYA–DACH developmental network (PSEDN) to better reflect the proteins involved [69]. This network plays a key regulatory role in the early development of multiple organs [70,71]. Notably, all known disease genes implicated in BORSD and OTFCS belong to this network. While OTFCS has also been genetically linked to PAX1 [4,14] and, in a limited number of patients, EYA4 in [19], EYA1 remains the major gene implicated in conditions.
Pathogenic EYA1 variants encompass truncating, missense, splice-site, stop-loss, and copy-number alterations, and have been documented in association with BORSD, OTFCS, and intermediate phenotypes [6,62]. Within our case series, one SNV in the EYA1 gene was described exclusively in association with OTFCS (1/141; 0.7%), one SNV was described in association with both OTFCS and BORSD (1/141; 0.7%), while the remainder (139/141; 98.6%) were described within the BORSD spectrum, with or without renal involvement. Thus, the variant class alone may be insufficient to predict the clinical presentation. This supports the view that haploinsufficiency is the predominant disease mechanism [72,73], but additional genetic or environmental modifiers likely influence phenotypic expressivity. Importantly, the observation that OTFCS can also result from missense and splice variants, and not exclusively from large deletions, further challenges the concept of OTFCS as a purely contiguous gene deletion syndrome [9,14]. Complex rearrangements, inversions, and insertions further contribute to the mutational spectrum [74,75].
A particularly noteworthy finding from our review is that the majority of published patients with OTFCS due to EYA1 defects presented with renal anomalies, while in approximately 25% of EYA1-related cases of BORSD, they were absent regardless of the variant type. Since renal involvement has been traditionally associated with BORSD, this observation undermines the concept of a strict clinical separation between the two syndromes. Instead, it suggests that musculoskeletal involvement in OTFCS and renal anomalies in BORSD are not mutually exclusive, but somewhat variable manifestations of the same allelic defect. Conversely, OTFCS caused by other genetic mechanisms may represent distinct subtypes, supporting the concept of a subclassification of OTFCS according to the underlying molecular etiology.
To our knowledge, this is the first reported case of OTFCS due to an EYA1 defect characterized by Chromosomal Microarray (CMA). This provides a precise genomic definition of the 8q13.2q13.3 microdeletion underlying the phenotype and allows a detailed assessment of co-deleted genes. We also acknowledge that the interpretation of microdeletion cases is complicated by the involvement of multiple genes. In our case, the 8q13.2q13.3 deletion spans additional genes besides EYA1. We therefore reviewed their known or predicted functions. Although some have roles in developmental pathways, none have been consistently implicated in renal or musculoskeletal phenotypes resembling BORSD or OTFCS. Thus, while we cannot completely rule out modifying effects from neighboring genes, the current evidence supports EYA1 haploinsufficiency as the principal driver of both the craniofacial–musculoskeletal anomalies and the renal phenotype in our patient.
The wide spectrum of presentations of EYA1-related disorders suggests that modifying factors, such as genetic background, environmental influences, or stochastic events during development, may critically modulate the expressivity of EYA1 variants [76]. Analogous patterns are well recognized in other genetic conditions such as COL2A1 (Collagen, Type II, Alpha-1)-related skeletal dysplasias and TBX6 (T-Box Transcription Factor 6)-related segmentation defects, where allelic heterogeneity and modifiers account for wide phenotypic variability [77,78,79,80]. Rather than being distinct syndromes, BORSD and OTFCS may represent different clinical expressions of EYA1 dysfunction within the context of the broader PSEDN. Reports of identical or highly similar EYA1 anomalies resulting in divergent phenotypes in different families further support this model [35,72,81].
From a clinical standpoint, acknowledging OTFCS and BORSD as allelic disorders has significant implications. It underscores the need to systematically evaluate musculoskeletal and developmental features in patients diagnosed with BORSD, and conversely, to ensure comprehensive renal and auditory assessments in patients with OTFCS. Grouping both under the umbrella of EYA1-related disorders would enhance and streamline variant interpretation, strengthen genetic counseling, and support the development of follow-up protocols that integrate renal, otologic, and skeletal surveillance.
Future studies should pursue three main directions: (i) large-scale genotype–phenotype analyses integrating both BORSD and OTFCS cases; (ii) functional studies to elucidate the molecular impact of different EYA1 variants; and (iii) investigation of potential second-site modifiers within the PSEDN Network that might influence phenotypic outcome.
6. Conclusions
Our findings consolidate the model of BORSD and OTFCS as allelic disorders within a unified EYA1-related spectrum. This reclassification is critical for clinical practice: it improves diagnostic accuracy, mandates comprehensive phenotyping—most notably, systematic renal screening in all OTFCS patients—and refines prognostic and genetic counseling. Future research integrating deep phenotyping, genomic data, and functional studies will be essential to elucidate the mechanisms underlying the striking phenotypic variability within this spectrum.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Johnson J.M. Moonis G. Green G.E. Carmody R. Burbank H.N. Syndromes of the First and Second Branchial Arches, Part 1: Embryology and Characteristic Defects Am. J. Neuroradiol.201132141910.3174/ajnr.A 207220299437 PMC 7964959 · doi ↗ · pubmed ↗
- 2Adams A. Mankad K. Offiah C. Childs L. Branchial Cleft Anomalies: A Pictorial Review of Embryological Development and Spectrum of Imaging Findings Insights Imaging 20167697610.1007/s 13244-015-0454-526661849 PMC 4729717 · doi ↗ · pubmed ↗
- 3Fára M. ChlupáckováV. Hrivnákova J. Familial oto-facio-cervical dysmorphia Acta Chir. Orthop. Traumatol. Cech.1967345115205585518 · pubmed ↗
- 4Gana S. Valetto A. Toschi B. Sardelli I. Cappelli S. Peroni D. Bertini V. Familial Interstitial 6q 23.2 Deletion Including Eya 4 Associated With Otofaciocervical Syndrome Front. Genet.20191065010.3389/fgene.2019.0065031379922 PMC 6656857 · doi ↗ · pubmed ↗
- 5Salinas-Torres V.M. Salinas-Torres R.A. Otofaciocervical Syndrome and Metachondromatosis in a Girl: Presentation of a Novel Association and Remarks on Clinical Variability of Branchial-Arch Disorders Int. J. Pediatr. Otorhinolaryngol.201685192110.1016/j.ijporl.2016.03.02127240490 · doi ↗ · pubmed ↗
- 6Castiglione A. Melchionda S. Carella M. Trevisi P. Bovo R. Manara R. Martini A. EYA 1-Related Disorders: Two Clinical Cases and a Literature Review Int. J. Pediatr. Otorhinolaryngol.2014781201121010.1016/j.ijporl.2014.03.03224803398 · doi ↗ · pubmed ↗
- 7Smith R.J. Branchiootorenal Spectrum Disorder Gene Reviews® Adam M.P. Feldman J. Mirzaa G.M. Pagon R.A. Wallace S.E. Amemiya A. University of Washington, Seattle Seattle, WA, USA 199320301554 · pubmed ↗
- 8Cho S.H. Jeong S.H. Choi W.H. Lee S.-Y. Genomic Landscape of Branchio-Oto-Renal Syndrome through Whole-Genome Sequencing: A Single Rare Disease Center Experience in South Korea Int. J. Mol. Sci.202425814910.3390/ijms 2515814939125727 PMC 11311636 · doi ↗ · pubmed ↗