SLP2/PHB Aggregates in ALS Mouse Models and Patients: Implications Beyond CHCHD10-Associated Motor Neuron Disease
Emmanuelle C. Genin, Françoise Lespinasse, Alessandra Mauri-Crouzet, Luc Dupuis, Véronique Paquis-Flucklinger

TL;DR
This study shows that SLP2/PHB aggregates appear in multiple forms of ALS, suggesting they may play a broader role in disease progression.
Contribution
The study expands the relevance of SLP2/PHB aggregates beyond CHCHD10-related ALS to other subtypes.
Findings
SLP2/PHB aggregates were found in FusΔNLS mice and some ALS patients, including those with C9ORF72 mutations.
Aggregates were absent in Sod1G86R mice and SOD1-associated ALS patients.
The presence of SLP2/PHB aggregates suggests a potential role in ALS pathogenesis across different genetic backgrounds.
Abstract
Amyotrophic Lateral Sclerosis (ALS) is a fatal neurodegenerative disorder characterized by motor neuron (MN) degeneration, frequently overlapping with frontotemporal dementia (FTD). Protein aggregation is a hallmark of these disorders, yet the role of aggregates in ALS pathogenesis remains unclear. Previously, stomatin-like protein 2 (SLP2) and prohibitin (PHB) aggregates were identified in a model of CHCHD10-related ALS (Chchd10S59L/+ mice). This study raises the question of the presence and possible involvement of these aggregates in ALS beyond CHCHD10-associated motor neuron disease (MND). Using immunohistofluorescence, we analyzed SLP2/PHB expression in the spinal MNs and hippocampus of two ALS mouse models: FusΔNLS and Sod1G86R. Additionally, post-mortem spinal cord tissues from 27 ALS and ALS-FTD patients were analyzed. SLP2/PHB aggregates were identified in spinal MNs and the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
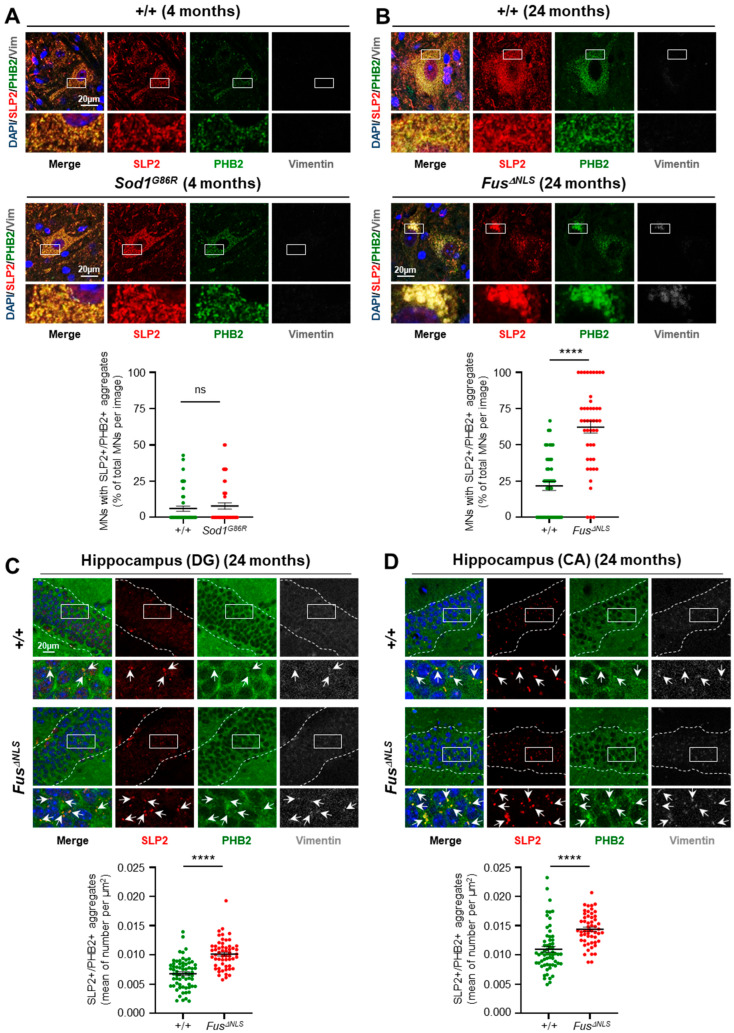 Figure 1
Figure 1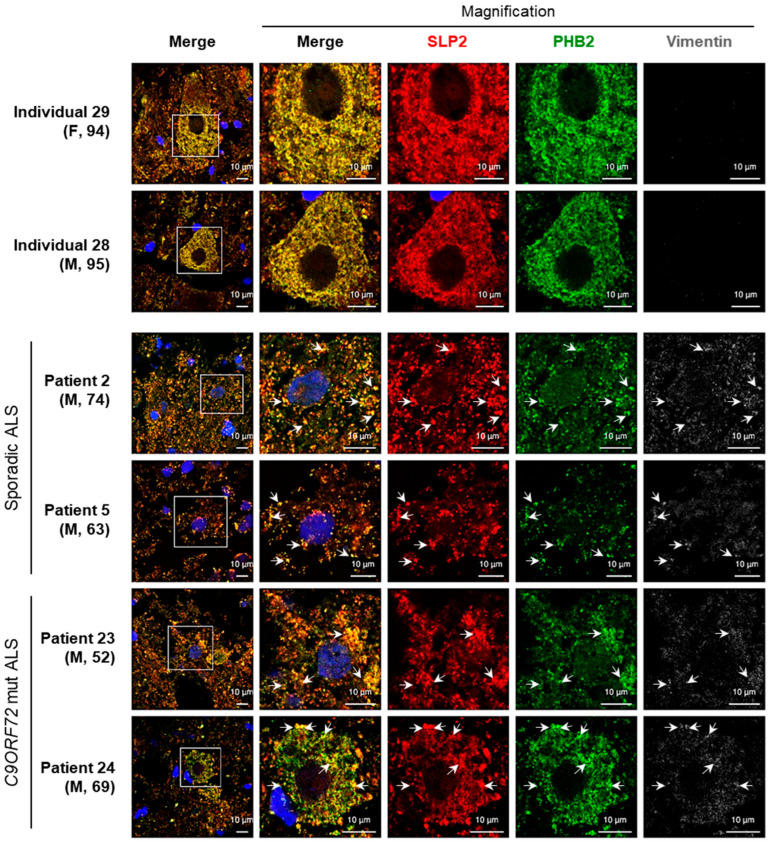 Figure 2
Figure 2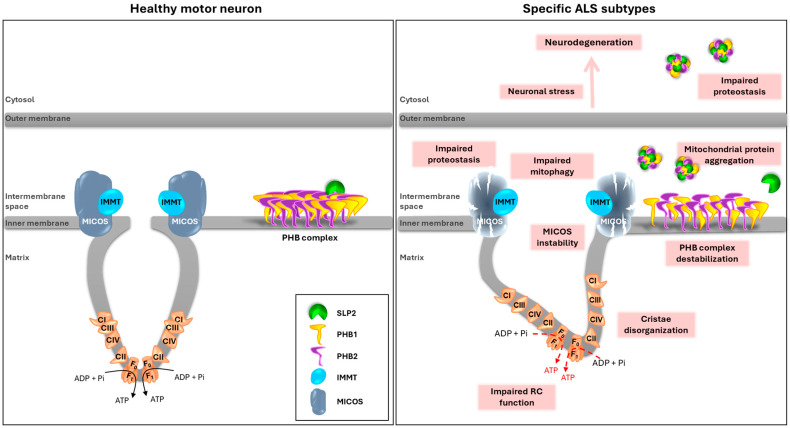 Figure 3
Figure 3- —Fondation pour la Recherche Médicale
- —Agence Nationale de la Recherche
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAmyotrophic Lateral Sclerosis Research · Genetic Neurodegenerative Diseases · Prion Diseases and Protein Misfolding
1. Introduction
Amyotrophic Lateral Sclerosis (ALS) is a fatal motor neuron disease (MND) characterized by the progressive degeneration of upper and lower motor neurons (MNs), ultimately leading to paralysis and death within 3 to 5 years after symptom onset [1]. ALS occurs in both sporadic (90–95%) and familial (5–10%) forms. The most frequent causative genes, responsible for more than 50% of familial ALS and about 7.5% of sporadic ALS, include C9ORF72 (Hexanucleotide repeat expansion in the chromosome 9 open reading frame 72), SOD1 (superoxide dismutase 1), TARDBP (encoding TDP-43, TAR DNA-binding protein 43), and FUS (Fused in Sarcoma) [2,3].
Clinically, ALS frequently overlaps with frontotemporal dementia (FTD), the second most common form of dementia after Alzheimer’s disease. ALS and FTD are now considered part of a continuous clinical spectrum: up to 50% of ALS patients display cognitive or behavioral symptoms indicative of frontal lobe involvement, while approximately 15% of FTD patients develop motor neuron signs. Shared variants in genes such as C9ORF72, TARDBP, and FUS further support the existence of overlapping pathogenic mechanisms [4,5]. FTD is a heterogeneous neurodegenerative disorder primarily affecting the frontal and anterior temporal lobes, leading to progressive impairments in cognition and behavior. To date, there is no effective disease-modifying treatments for ALS, FTD, or ALS-FTD, highlighting the need for a deeper understanding of the molecular pathways involved to identify new therapeutic strategies [6].
ALS and FTD also share neuropathological hallmarks, notably the accumulation of misfolded proteins forming cytoplasmic inclusions in degenerating neurons. These proteinopathies, including aggregates of TDP-43, SOD1, and FUS, highlight crucial dysregulation in protein homeostasis and cellular stress responses [7,8]. More recently, novel mitochondria-localized protein aggregates have emerged as potential contributors to disease mechanisms. Notably, aggregates of CHCHD10, a mitochondrial protein, have been described in the brain of FTD patients [9] as well as in cellular and mouse models expressing CHCHD10 mutations [9,10,11,12,13,14,15].
Among the genetic forms of ALS-FTD, CHCHD10 has been identified as a causative gene (ALS-FTD2, OMIM #615911), particularly in patients harboring the heterozygous p.Ser59Leu (S59L) variant [16,17,18,19,20]. CHCHD10 encodes a mitochondrial intermembrane protein enriched at cristae junctions, where it contributes to mitochondrial structure and function. The identification of CHCHD10 variants in ALS-FTD patients provided the first genetic evidence directly linking primary mitochondrial disfunction to MN degeneration. To investigate the pathogenic mechanisms associated with CHCHD10 variants, we previously developed a Chchd10^S59L/+^ knock-in mouse model, which recapitulates key clinical and pathological features of ALS-FTD [10,11]. These mice exhibit neuromuscular junction dysfunction, spinal MN degeneration, and TDP-43 cytoplasmic aggregates, consistent with the TDP-43 proteinopathy observed in ALS patients [11,12]. They also display hippocampal protein inclusions and cognitive impairments resembling FTD [10]. Notably, CHCHD10 aggregates are observed in both spinal and hippocampal regions, some of which colocalize with phosphorylated TDP-43 aggregates [9,10].
Previously, we demonstrated that CHCHD10 interacts with stomatin-like protein 2 (SLP2) [15], a mitochondrial scaffolding protein that stabilizes the prohibitin (PHB) complex, composed of PHB1 and PHB2, in the inner mitochondrial membrane [21]. In Chchd10^S59L/+^ mice, we observed the formation of perinuclear SLP2/PHB aggregates in spinal MNs and smaller aggregates in the hippocampus [10,15]. These aggregates correlate with neuronal death, suggesting that abnormal clustering of mitochondrial scaffold proteins may contribute to neurodegeneration in CHCHD10-associated ALS-FTD [15]. However, whether mitochondrial SLP2/PHB aggregation represents a common pathological mechanism across ALS subtypes, beyond *CHCHD10-*related disease, remains unknown. This represents a critical knowledge gap in understanding the broader role of mitochondrial dysfunction in ALS pathogenesis.
Here, we investigated whether mitochondrial scaffold protein aggregation involving SLP2 and PHBs constitutes a shared pathological hallmark across genetically diverse ALS models and patient tissues, potentially uncovering novel biomarkers or therapeutic targets relevant to precision medicine approaches. To address this, we analyzed the presence of SLP2/PHB aggregates in two well-established ALS mouse models with distinct pathogenic mechanisms: the Sod1^G86R^ transgenic model, which mimics toxic gain-of-function effects of mutant SOD1 and is characterized by mitochondrial stress and oxidative damage [22,23]; and the Fus^ΔNLS^ knock-in model, which presents a defective nuclear localization signal, leading to cytoplasmic FUS accumulation and downstream TDP-43 pathology [24,25]. These complementary models encompass both mitochondrial and RNA-binding protein-related mechanisms of MND, providing an ideal framework to determine whether SLP2/PHB aggregation is a common feature of ALS, independent of its genetic origin.
Finally, to assess the translational relevance of our findings, we extend our analysis to postmortem tissues from 27 ALS and ALS-FTD patients. Our results reveal that SLP2/PHB aggregates are also present in human ALS cases, regardless of genetic background. These findings suggest that the aggregation of mitochondrial scaffold protein represents a more general pathogenic mechanism in ALS than previously thought, with potential implications for diagnostic and therapeutic strategies.
2. Results
2.1. SLP2/PHB Aggregation in FusΔNLS Mice but Not in the Sod1G86R Model
Our research on Chchd10^S59L/+^ mice demonstrated that SLP2 and PHB aggregates accumulate in degenerating spinal MNs and hippocampal neurons, correlating with neuronal loss [10,15]. This led us to investigate whether these aggregates are exclusive to CHCHD10-associated disease or found in ALS of other origins. To this end, we analyzed SLP2/PHB aggregates in two ALS mouse models: Sod1^G86R^, which exhibits a severe form as early as 4 months of age [22,23], and Fus^ΔNLS^, which develops a late-onset disease with symptoms at 24 months of age [24,25]. Contrary to our results in Chchd10^S59L/+^ mice at the end-stage (around one year of age) [15], immunostaining for SLP2 and PHB2 did not reveal any abnormal expression in the spinal MNs of Sod1^G86R^ mice at end-stage (4 months) (Figure 1A). However, large perinuclear aggregates expressing both SLP2 and PHB2 were observed in spinal MNs of Fus^ΔNLS^ mice at 24 months of age (Figure 1B). These aggregates were also positive for vimentin, consistent with their aggresome-related nature. Quantitative analysis confirmed a significant increase in the proportion of MNs containing SLP2/PHB aggregates in Fus^ΔNLS^ mice compared to controls. Furthermore, significant SLP2 and PHB2 aggregation was detected in Fus^ΔNLS^ mice hippocampus (Figure 1C,D), mirroring the patterns observed in Chchd10^S59L/+^ mice [15].
2.2. Aggregates Including SLP2 and PHB2 Are Also Present in ALS Patients
To extend the relevance of these findings to human disease, we analyzed a series of 27 patients diagnosed with ALS and ALS-FTD. This cohort included 14 individuals with sporadic ALS, aged between 54 and 74 years, 2 with sporadic ALS-FTD (aged 42 and 70), and 11 patients presenting with monogenic forms of ALS. Among the latter, there were 7 with C9ORF72 mutations (5 diagnosed with ALS and 2 with ALS-FTD, aged 53 to 77), 3 with SOD1 mutations (aged 50 to 73), and 1 patient with TARBDP-associated disease (aged 47) (Table 1). Additionally, we analyzed 3 control subjects (aged 84, 94, and 95), two of whom exhibited Braak stage III Alzheimer’s lesions.
We identified SLP2/PHB2 aggregates in spinal MNs of 2 patients with sporadic ALS (patients 2 and 5, aged 74 and 63, respectively) and 2 patients presenting a C9ORF72-associated ALS (patients 23 and 24, aged 52 and 69, respectively) (Figure 2). Quantitative analysis revealed a significant increase in the proportion of MNs containing SLP2/PHB aggregates in ALS patients compared to controls (Table 2). In sporadic ALS patients, 43–45% of MNs contained aggregates vs. 14–18% in controls. Similarly, in C9ORF72-associated ALS patients, 39–48% of MNs harbored aggregates, confirming a significant enrichment relative to control individuals. Notably, as for the Sod1^G86R^ mouse model, these aggregates were absent in patients with SOD1 variants. Taken together, these results suggest that SLP2/PHB aggregation is not specific to CHCHD10-associated disease and that SLP2 and prohibitins may be involved in the cascade of events leading to neuronal death in ALS of other origins.
3. Discussion
This study provides the first evidence that SLP2/PHB protein aggregation may contribute to ALS pathogenesis beyond CHCHD10 mutations, revealing a potentially broader role for mitochondrial scaffold protein dysregulation in MND (Figure 3).
3.1. SLP2/PHB Aggregation in the FusΔNLS ALS Model
In the Fus^ΔNLS^ knock-in mouse model, we identified large perinuclear SLP2/PHB aggresomes in spinal MNs and smaller aggregates in the hippocampus. Quantification revealed an approximately 190% increase in the spinal cord, 50% increase in the DG, and 30% increase in the CA region of Fus^ΔNLS^ mice compared to controls. These findings closely mirror those previously observed in Chchd10^S59L/+^ mouse model, where SLP2 and PHB form aggregates correlating with neurodegeneration, showing an approximate 210% increase in the spinal cord, 166% in DG, and 94% in CA at the end-stage of disease [10,15]. Importantly, the occurrence of such aggregates in Fus^ΔNLS^ mice, harboring a mutation unrelated to mitochondrial genes, suggests that SLP2/PHB aggregation may be a downstream consequence of convergent neurodegenerative mechanisms, rather than a feature exclusive to CHCHD10-related disease.
3.2. Detection of SLP2/PHB Aggregates in ALS Patient Tissues
Consistent with this, we observed SLP2/PHB aggregates in spinal motor neurons of 4 ALS out of 27 ALS and ALS-FTD patients. Quantitative analysis showed that control individuals exhibited ~17.7% and ~14.3% of MNs containing SLP2/PHB aggregates, whereas two sporadic ALS patients displayed ~43.5% and ~45.1%, and two C9ORF72 ALS patients showed ~47.6% and ~39.5% of MNs with SLP2/PHB aggregates. Although the morphology of these aggregates differed from those observed in mouse models, probably due to post-mortem tissue degradation, fixation artifacts, or disease stage, their presence in human tissues supports the clinical relevance and translational potential of our findings. These observations reinforce the hypothesis that mitochondrial scaffold protein aggregation may contribute to disease pathology in a subset of ALS patients, independent of the underlying genetic cause.
3.3. Absence of SLP2/PHB Aggregates in the Sod1G86R ALS Model
Interestingly, no SLP2/PHB aggregates were observed in the Sod1^G86R^ mouse model. This result is particularly relevant since we specifically selected the G86R variant, which (unlike the widely used G93A mutant) does not accumulate in mitochondria upon overexpression [26], thereby minimizing potential confounding effect related to abnormal mitochondrial localization of SOD1. The absence of SLP2/PHB aggregates in Sod1^G86R^ mice, combined with their presence in only a minority of human ALS samples, suggests that SLP2/PHB aggregation is not a universal feature of ALS, but rather a pathological hallmark of specific molecular subtypes. This aligns with the known clinical and pathological heterogeneity of ALS; wherein different patients exhibit distinct proteinopathy profiles and cellular stress responses [27,28]. These findings highlight the importance of molecularly stratified approaches in both the study and treatment of ALS.
3.4. Age-Dependent Accumulation of SLP2/PHB Aggregates
Our data also suggest that SLP2/PHB aggregation may accumulate progressively with aging in mouse models. In Chchd10^S59L/+^ mice, SLP2/PHB aggregates are absent at early stages (e.g., 3 months of age) but become increasingly prominent with age [15]. This progressive accumulation may reflect the gradual deterioration of mitochondrial function, impaired proteostasis, and reduced autophagic clearance, hallmarks of aging known to contribute to neurodegenerative processes [29,30,31]. In particular, the formation of SLP2/PHB aggregates may indicate a failure of mitophagy, the selective autophagic clearance of damaged mitochondria, which is increasingly recognized as a critical pathway disrupted in ALS and other neurodegenerative disorders [32,33]. Impaired autophagic flux or overload of the degradation system could contribute to the persistence and accumulation of these mitochondrial aggregates, further exacerbating neuronal stress and degeneration. Notably, we also detected these aggregates in a small number of MNs in aged control mice, further supporting the idea that aging may independently promote their formation. In human samples, however, the presence of SLP2/PHB aggregates was not restricted to the oldest individuals in the cohort (aged 52, 63, 69, and 74 years), suggesting that factors beyond chronological age, such as genetic predisposition or cumulative cellular stress, may influence aggregate formation.
3.5. Integrative Model of SLP2/PHB Aggregation in ALS Pathogenesis
Altogether, these results point to SLP2/PHB aggregation as a multifactorial phenomenon, influenced by both genetic mutations (e.g., CHCHD10, FUS, C9ORF72) and age-related cellular decline. The formation of these aggregates may represent a converging pathological mechanism affecting mitochondrial function, possibly through disruption of mitochondrial architecture, cristae dynamics, or inner membrane homeostasis. Moreover, our findings raise the possibility that SLP2/PHB aggregation reflects impaired mitophagic processes in certain ALS subtypes. Given the central role of autophagy in maintaining mitochondrial quality control, especially in post-mitotic neurons, future studies should explore whether modulating autophagic activity could prevent or reduce the accumulation of these aggregates. Targeting autophagy or mitophagy-related pathways may therefore represent a promising therapeutic avenue for ALS subtypes characterized by mitochondrial proteinopathy [34,35].
4. Materials and Methods
4.1. Mouse Models
Sod1^G86R^ and Fus^ΔNLS^ mice were generated as described in [24,25] and housed at Faculty of Medicine from Strasbourg University under controlled conditions (12/12 h light/dark cycle, 21 ± 1 °C, 60% humidity) with ad libitum access to food and water. Fus^ΔNLS^ mice, expressing a truncated FUS protein lacking the PY-NLS (encoded by exon 15 of the Fus gene), were maintained in congenic C57Bl6/J. Sod1^G86R^ mice (FVB-Tg(Sod1*G86R)M1Jwg/J, Jax Strain#005110) were maintained in a pure FVB/N background. To reduce animal use, tissues from previous studies were used [36,37]. All experiments were approved by the Strasbourg University ethical committee (CREMEAS, references 2016111716439395 and 25452). No inclusion or exclusion criteria were applied for animal selection, and no data points were excluded during analysis.
4.2. Immunohistofluorescence on Mouse Tissues
Immunostaining was performed on lumbar spinal cord sections from Sod1^G86R^ and Fus^ΔNLS^ mice and on brain sections from Fus^ΔNLS^ mice as previously described [15]. Briefly, antigen retrieval was performed in citrate buffer (pH6.0; Vector Laboratories, H3300) for 15 min at 95 °C followed by 15 min at room temperature (RT). Brain or spinal cord sections were then incubated for 1 h at RT in blocking buffer containing 0.25–0.5% Triton X-100, 5% normal goat serum, and 4% bovine serum albumin (BSA). Floating sections were incubated overnight at 4 °C with primary antibodies diluted in blocking buffer: mouse anti-PHB2 (Proteintech, Rosemont, IL, USA, 66424-1-Ig; 1/400), rabbit anti-SLP2 (Proteintech, 10348-1-AP; 1/100), and goat anti-Vimentin (Merck, Darmstadt, Germany, V4360; 1/150). After several washes, sections were incubated for 1 h at RT in a solution of PBS containing 0.3% Triton with the corresponding Alexa Fluor-conjugated secondary antibodies: Alexa Fluor 488 (Invitrogen, (Thermo Fisher Scientific) Waltham, MA, USA, A11029; 1/500), Alexa Fluor 594 (Invitrogen, A21207; 1/500), and Alexa Fluor 647 (Invitrogen, A32849; 1/500). Nuclei were counterstained DAPI (2 µg/mL; Life Technologies (Thermo Fisher Scientific), Carlsbad, CA, USA) for 5 min at RT. Sections were then mounted using Vectashield Hard Set medium (Vector Laboratories, Newark, CA, USA).
Confocal images were acquired using a ZEISS LSM880 laser-scanning microscope. Aggregate quantification was performed using FIJI (ImageJ 1.54p; National Institutes of Health, Bethesda, MD, USA). Manual annotation of MNs containing SLP2/PHB aggregates was performed in a blinded manner with respect to genotype.
4.3. Immunohistofluorescence on Human ALS Tissues
Immunostaining on human lumbar spinal cord tissues was performed on 15 µm sections from 30 individuals, including 27 ALS patients and 3 controls (Table 1). Sections were fixed in 4% paraformaldehyde for 20 min, followed by antigen retrieval in 0.01M citrate buffer (pH 6) at 90 °C for 12 min. Samples were then permeabilized and blocked for 1 h in PBS containing 0.25% Triton X-100 (Sigma-Aldrich, Darmstadt, Germany), 4% bovine serum albumin, and 5% normal goat serum. Immunolabelling was performed using the same antibodies and dilutions as for mouse tissues.
To mitigate lipofuscin autofluorescence, sections were mounted using EverBrite™ TrueBlack^®^ Hardset mounting medium (Biotium, Fremont, CA, USA, #23018-T). Imaging was performed with a confocal laser scanning microscope (LSM880; Carl Zeiss, Oberkochen, Germany) to assess SLP2 and PHB expression. Manual annotation of MNs containing SLP2/PHB aggregates was performed in a blinded manner. Human spinal cord samples were provided from the Brainbank Neuro-CEB Neuropathology Network and the Pitié-Salpétrière Hospital, (Paris, France).
4.4. Statistical Analysis
Quantitative analysis of SLP2/PHB aggregates in lumbar spinal MNs of Sod1^G86R^ and Fus^ΔNLS^ mice, as well as in brain sections from Fus^ΔNLS^ mice, was performed using GraphPad Prism 8.0 (GraphPad Software). Comparisons between two independent groups were conducted using the non-parametric Mann–Whitney U test. All analyses were two-tailed, and p-values < 0.05 were considered statistically significant.
For human spinal cord tissues, statistical analyses were performed on 4 ALS patients exhibiting SLP2/PHB aggregates in >30% of analyzed MNs and 2 control individuals (17–51 MNs per individual). The frequency distribution of MNs with (pathological) versus without (non-pathological) SLP2/PHB aggregates was compared between ALS patients and controls using the chi-square (χ^2^) test in GraphPad Prism 8.0 (GraphPad Software). p-values < 0.05 were considered statistically significant.
5. Conclusions
In conclusion, these results suggest that SLP2/PHB aggregation not only may reflect a structural disruption of mitochondrial homeostasis but may also be indicative of impaired mitochondrial turnover, potentially involving autophagy and mitophagy dysfunctions. Our findings expand the spectrum of proteinopathy in ALS to include mitochondrial scaffold protein aggregation, offering new insights into disease pathogenesis and highlighting potential therapeutic targets. Future work should aim to elucidate the molecular mechanisms by which SLP2 and PHB contribute to neuronal dysfunction and determine whether modulating their expression or preventing their aggregation could ameliorate mitochondrial dysfunction and neurodegeneration in ALS. Additionally, broader studies in genetically diverse patient cohorts will be essential to define the prevalence and clinical relevance of SLP2/PHB aggregation and to explore its utility as a biomarker for precision medicine approaches in ALS.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Masrori P. Van Damme P. Amyotrophic Lateral Sclerosis: A Clinical Review Eur. J. Neurol.2020271918192910.1111/ene.1439332526057 PMC 7540334 · doi ↗ · pubmed ↗
- 2Genin E.C. Abou-Ali M. Paquis-Flucklinger V. Mitochondria, a Key Target in Amyotrophic Lateral Sclerosis Pathogenesis Genes 202314198110.3390/genes 1411198138002924 PMC 10671245 · doi ↗ · pubmed ↗
- 3Rizea R.E. Corlatescu A.-D. Costin H.P. Dumitru A. Ciurea A.V. Understanding Amyotrophic Lateral Sclerosis: Pathophysiology, Diagnosis, and Therapeutic Advances Int. J. Mol. Sci.202425996610.3390/ijms 2518996639337454 PMC 11432652 · doi ↗ · pubmed ↗
- 4Parobkova E. Matej R. Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degenerations: Similarities in Genetic Background Diagnostics 20211150910.3390/diagnostics 1103050933805659 PMC 7998502 · doi ↗ · pubmed ↗
- 5Van Es M.A. Hardiman O. Chio A. Al-Chalabi A. Pasterkamp R.J. Veldink J.H. Van Den Berg L.H. Amyotrophic Lateral Sclerosis Lancet 20173902084209810.1016/S 0140-6736(17)31287-428552366 · doi ↗ · pubmed ↗
- 6Petrov D. Mansfield C. Moussy A. Hermine O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment?Front. Aging Neurosci.201796810.3389/fnagi.2017.0006828382000 PMC 5360725 · doi ↗ · pubmed ↗
- 7Benson B.C. Shaw P.J. Azzouz M. Highley J.R. Hautbergue G.M. Proteinopathies as Hallmarks of Impaired Gene Expression, Proteostasis and Mitochondrial Function in Amyotrophic Lateral Sclerosis Front. Neurosci.20211578362410.3389/fnins.2021.78362435002606 PMC 8733206 · doi ↗ · pubmed ↗
- 8Wilson D.M. Cookson M.R. Van Den Bosch L. Zetterberg H. Holtzman D.M. Dewachter I. Hallmarks of Neurodegenerative Diseases Cell 202318669371410.1016/j.cell.2022.12.03236803602 · doi ↗ · pubmed ↗