Integrated Enzyme-Mediated One-Step Sample Processing and Duplex Amplification System for Rapid Detection of Carpione rhabdovirus in Aquaculture-Derived Food Products
Heng Sun, Haoyu Wang, Jie Huang, Yao Wu, Zhenxin Hu, Yucong Huang

TL;DR
A new rapid detection system for a virus affecting golden pompano in aquaculture uses enzyme-based methods to improve speed and accuracy.
Contribution
The novel EmOSP-RT-EmDEA system integrates sample processing and amplification for rapid CAPRV detection without exogenous probes.
Findings
The system detects CAPRV2023 with a sensitivity of 4 copies/μL and matches TaqMan qPCR performance in fecal samples.
It enables detection across clinical, invasive, and environmental specimens with high specificity.
The EmOSP protocol provides robust and reproducible nucleic acid extraction from fecal, hepatic, and water samples.
Abstract
Golden pompano (Trachinotus ovatus) is the largest-scale marine aquaculture fish species in China, with a significant economic and nutritional value as a high-quality seafood product. The recent outbreak of an epidemic caused by a novel Carpione rhabdovirus (CAPRV) occurred in cultured golden pompano. To address it, a CAPRV enzyme-mediated one-step sample processing–reverse transcription–enzyme-mediated duplex exponential amplification (EmOSP-RT-EmDEA) detection system was developed. This innovative molecular diagnostic tool integrates enzyme-mediated one-step sample processing (EmOSP) with enzyme-mediated duplex exponential amplification (EmDEA) technology. Unlike traditional RPA-Cas12a detection methods, this system directly incorporates fluorophores into RNA components, eliminating the need for exogenous fluorescent probes while maintaining high sensitivity. It enables rapid,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
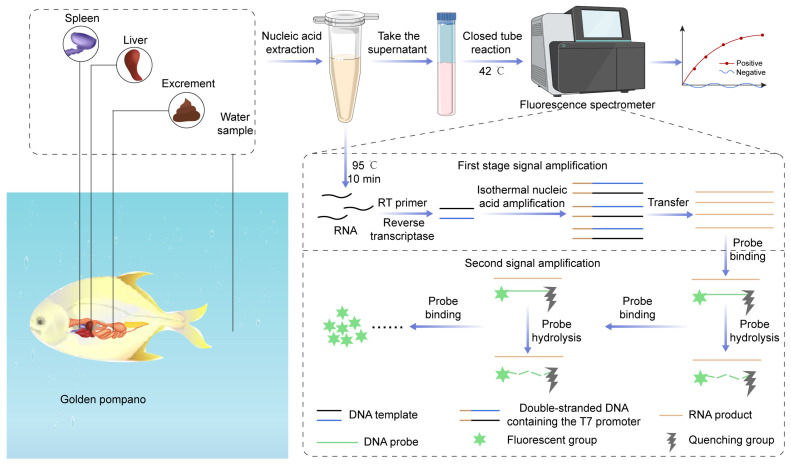 Figure 1
Figure 1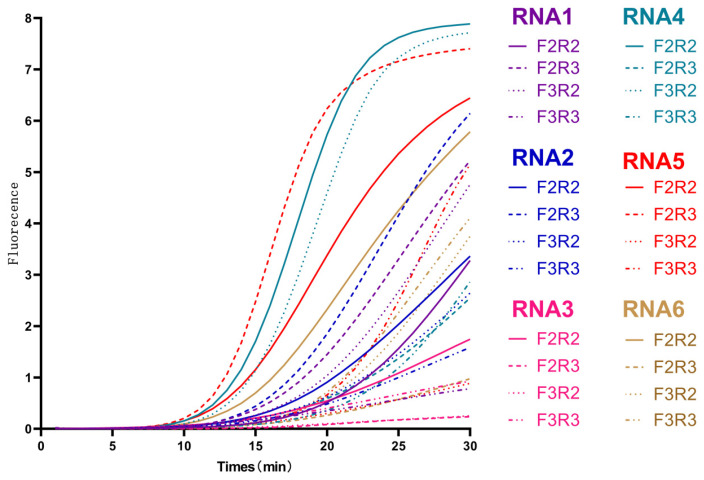 Figure 2
Figure 2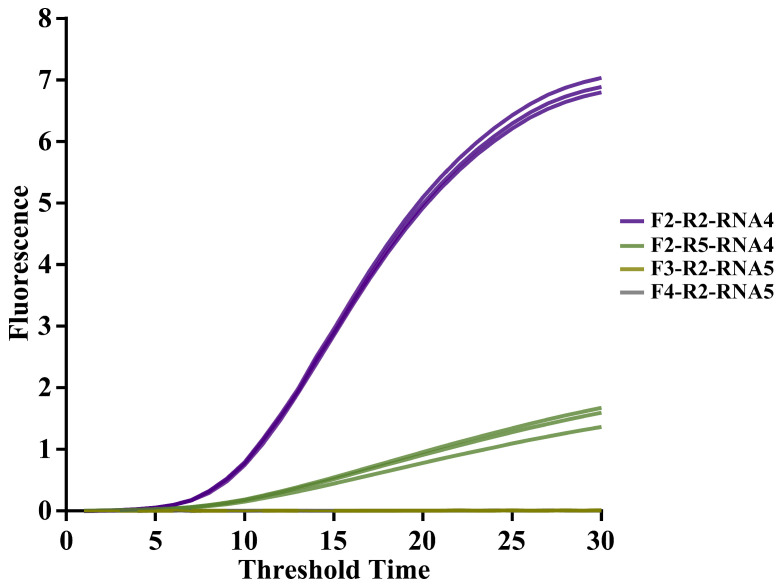 Figure 3
Figure 3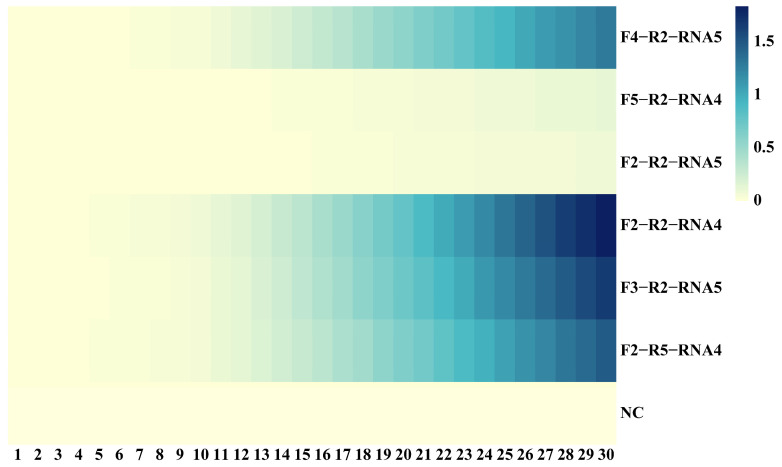 Figure 4
Figure 4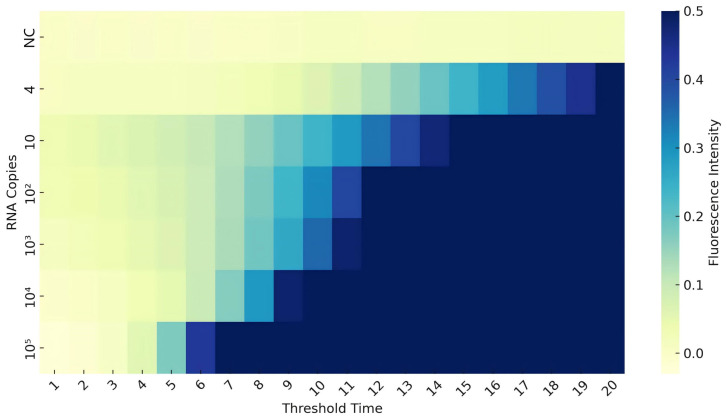 Figure 5
Figure 5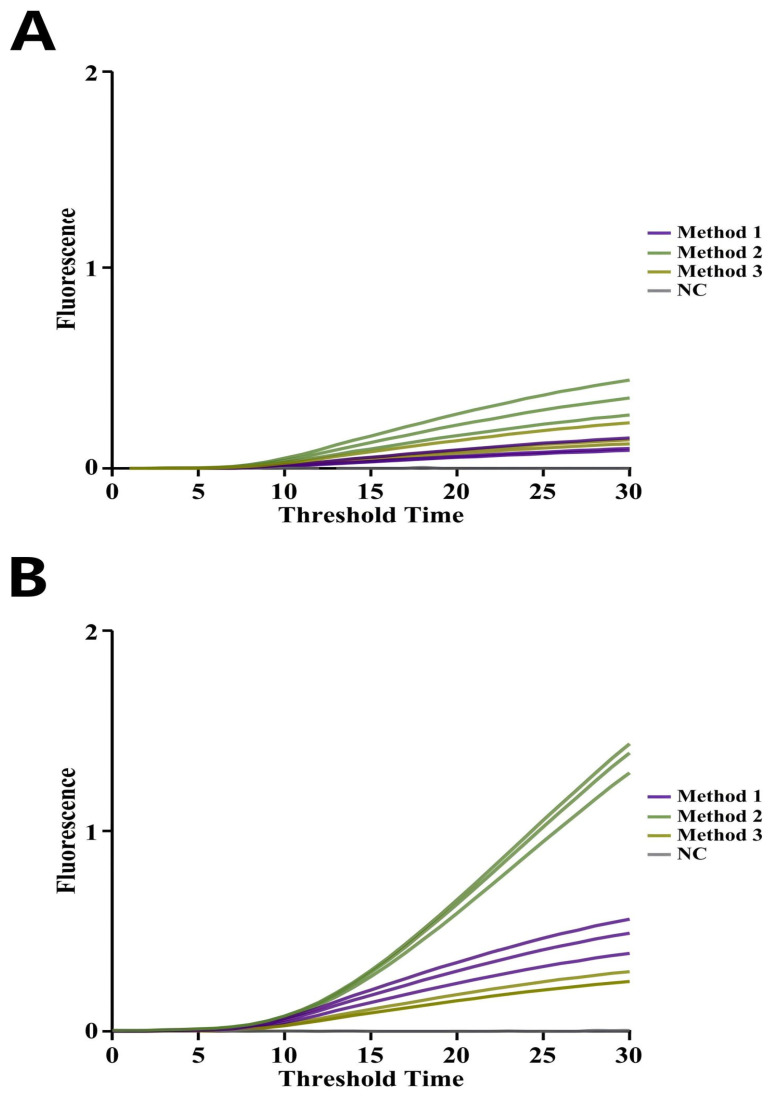 Figure 6
Figure 6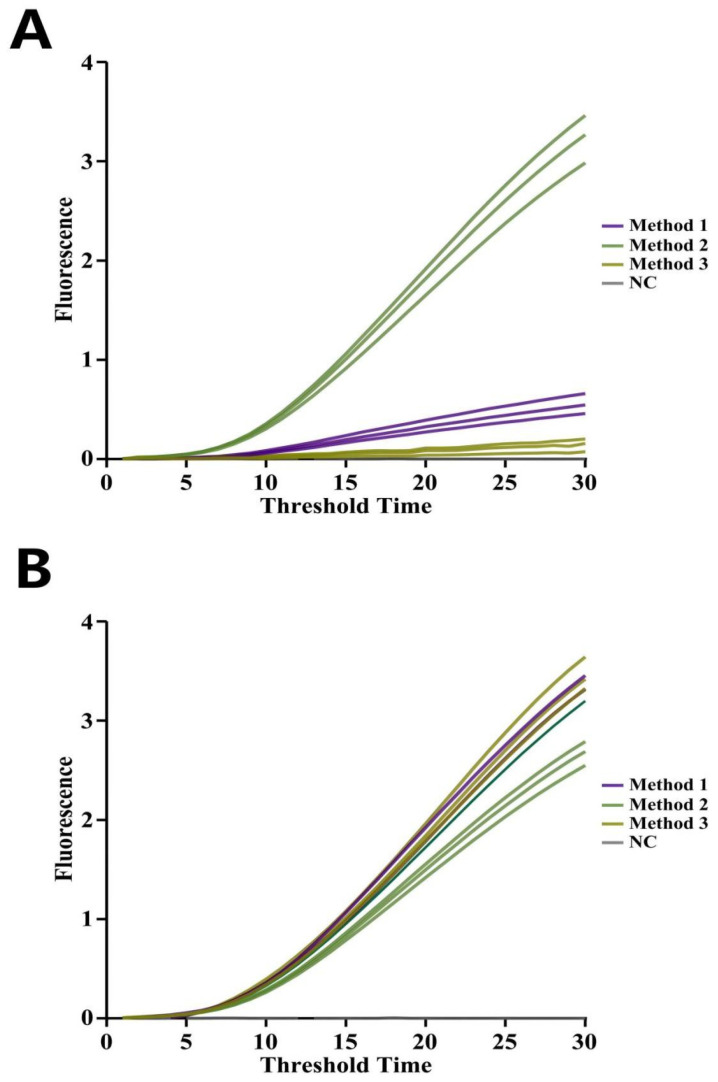 Figure 7
Figure 7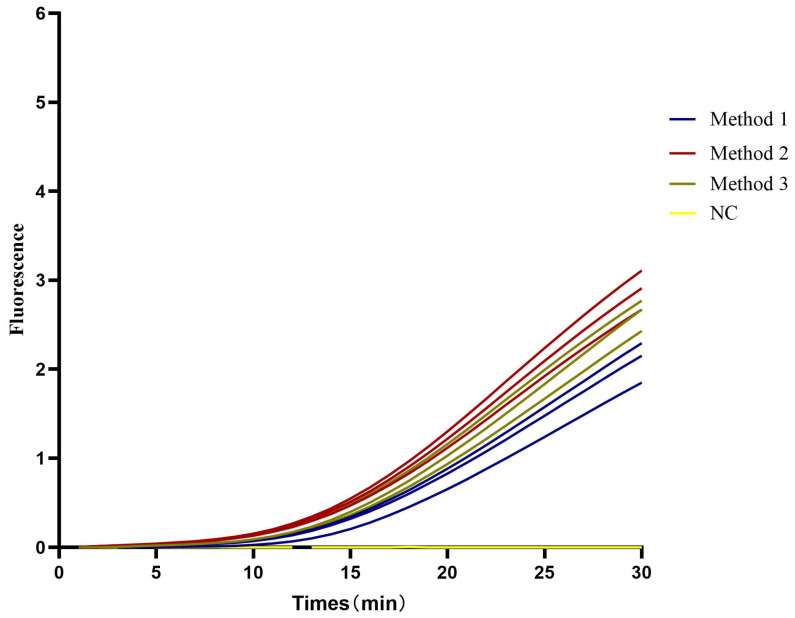 Figure 8
Figure 8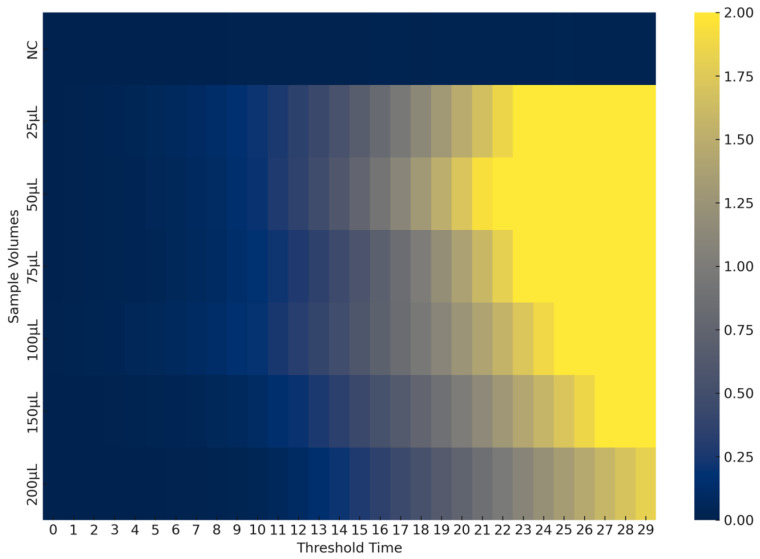 Figure 9
Figure 9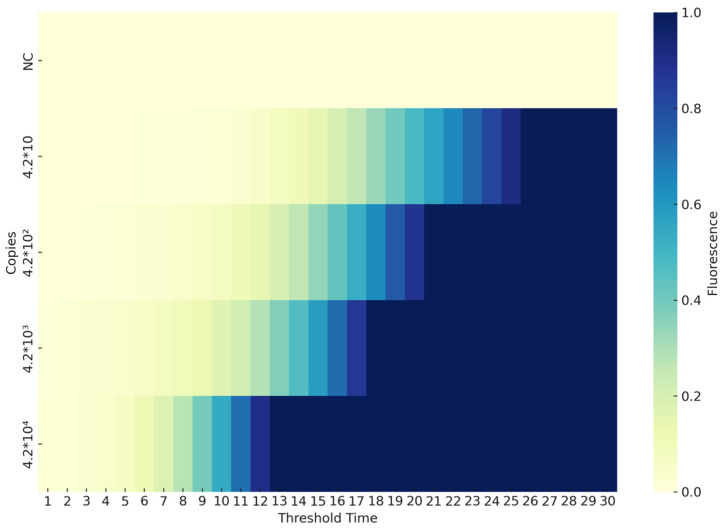 Figure 10
Figure 10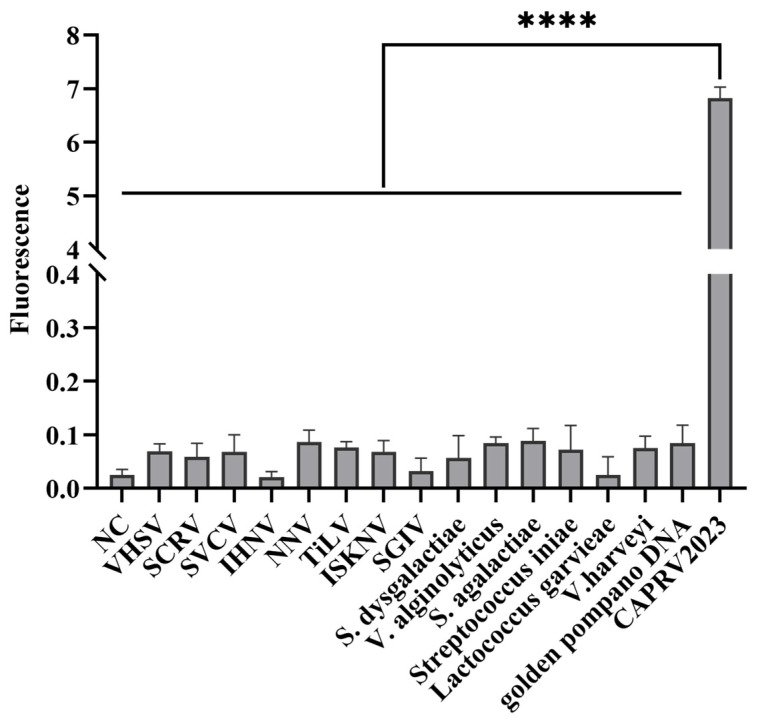 Figure 11
Figure 11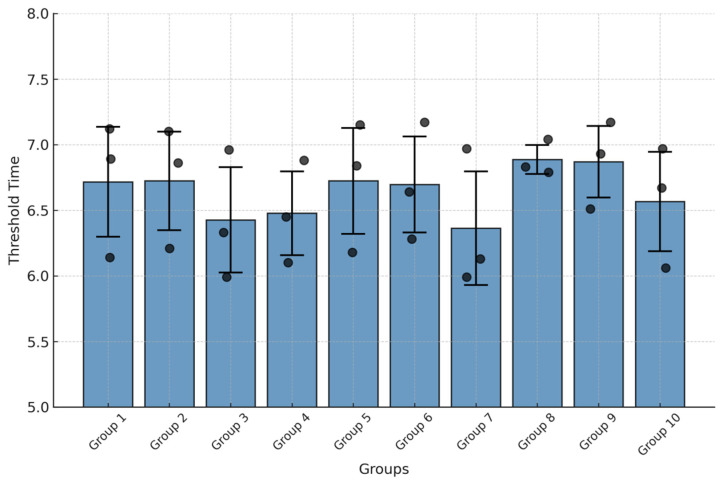 Figure 12
Figure 12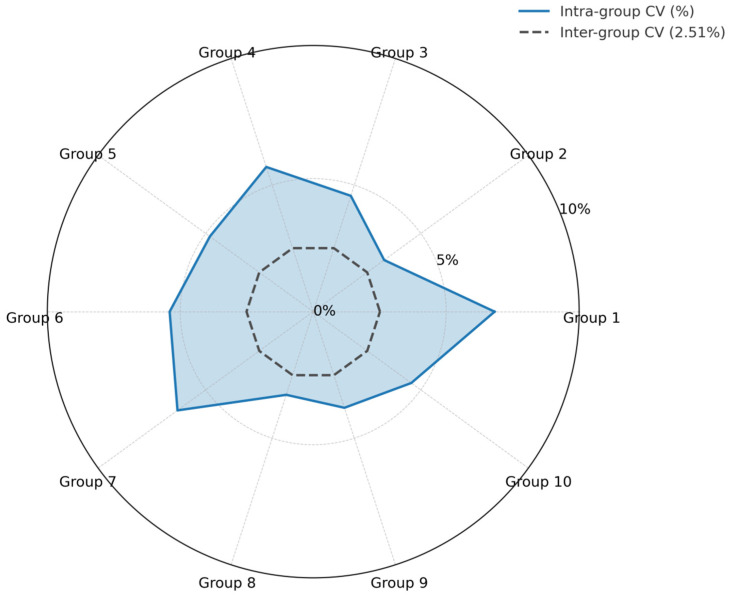 Figure 13
Figure 13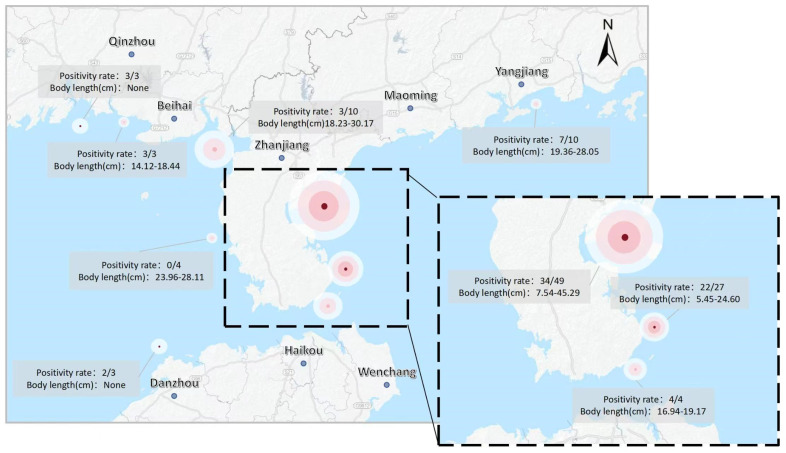 Figure 14
Figure 14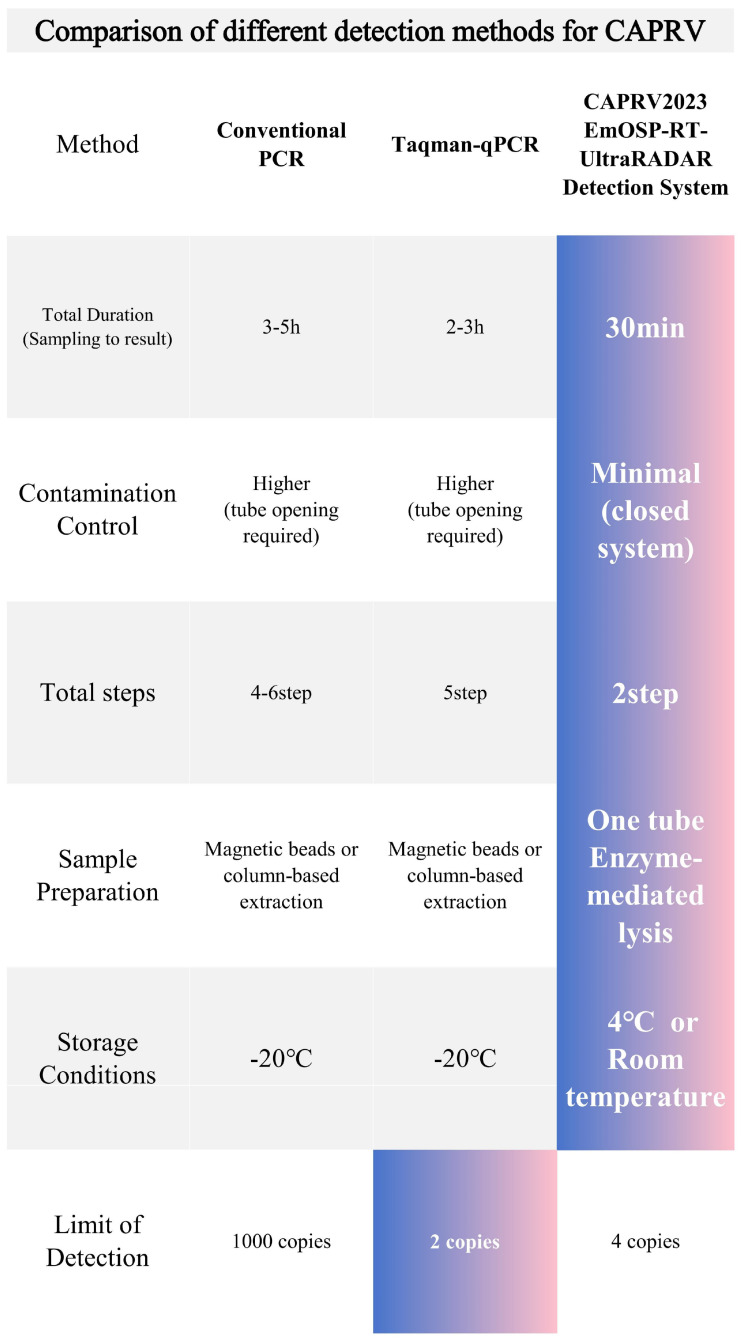 Figure 15
Figure 15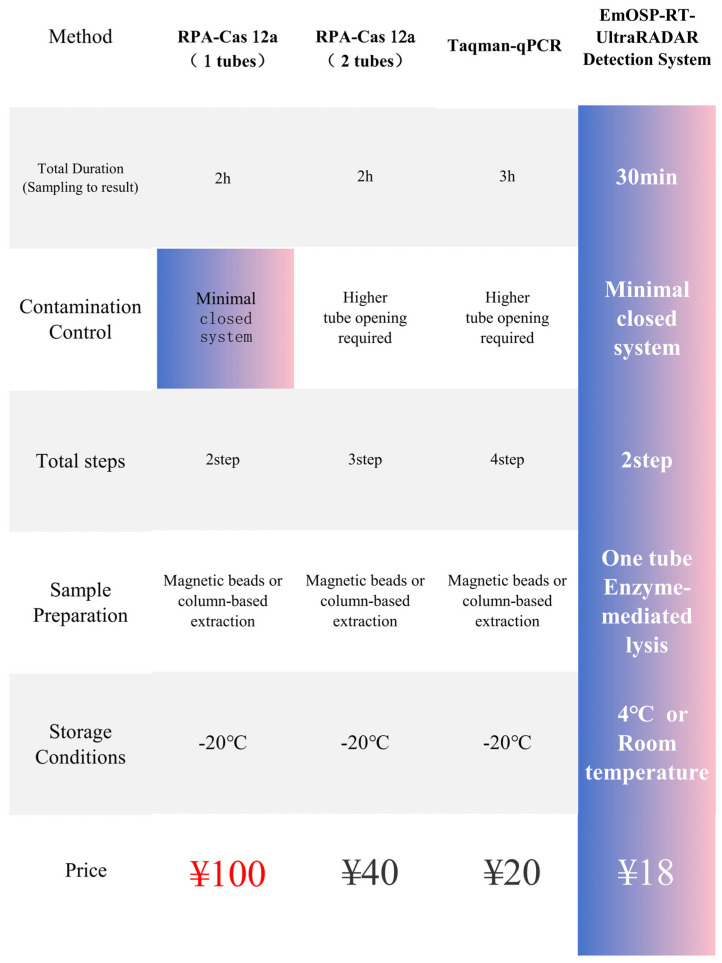 Figure 16
Figure 16- —Key Scientific Research Platforms and Projects of Regular Institutions of Higher Education in Guangdong Province
- —Innovation Program in Postgraduate Education of Guangdong Ocean University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsIdentification and Quantification in Food · Aquaculture disease management and microbiota · Molecular Biology Techniques and Applications
1. Introduction
Trachinotus ovatus, commonly known as golden pompano, is a species in the genus Trachinotus of the Carangidae family, widely distributed in subtropical and tropical seawaters of Southeast Asia. In recent years, due to its rapid growth, strong adaptability, and high economic value, golden pompano has become the most widely cultivated fish species in net cage aquaculture along China’s southern coast. According to data from the China Fisheries Statistical Yearbook 2024, the cultured production of golden pompano in China reached 292,263 tons in 2023 [1,2,3,4].
In the autumn of 2023, a novel acute infectious disease emerged among cage-farmed golden pompano in southern China, resulting in mortality rates ranging from 65% to 82% within just 7 to 10 days. This outbreak posed a significant threat to China’s marine aquaculture industry [1,2]. A novel rhabdovirus strain was isolated from the infected golden pompano and designated as Carpione rhabdovirus 2023 (CAPRV2023) [1,2]. CAPRV virions are enveloped, bullet-shaped particles (approximately 183–218 × 57–83 nm) containing a negative-sense single-stranded RNA genome (~11.7 kb) with the canonical 3′-N-P-M-G-NV-L-5′ gene organization, characteristic of the genus Novirhabdovirus. CAPRV2023 represents only the second recorded strain of this viral species, following the initial isolation of strain CAPRV583 in Italy from the carpione (Salmo trutta carpio) [1,2,3]. Notably, the whole-genome similarity between CAPRV2023 and CAPRV583 is only 81.32% [1,2,3].
Due to the lack of antiviral drugs and corresponding vaccines, there are currently no effective control measures for this emerging infectious disease. Therefore, there is an urgent need to develop precise and sensitive molecular assays to detect CAPRV2023 as early and accurately as possible. Early detection is crucial for enabling effective prevention and control measures. To date, in addition to traditional necropsy and pathological examination, Sun et al. have developed various diagnostic methods for detecting CAPRV2023, including cell culture, in situ hybridization, electron microscopy, reverse transcription polymerase chain reaction (RT-PCR) and SYBR Green I quantitative PCR (qPCR) [1,2,3].
Recombinase polymerase amplification (RPA) is an emerging isothermal nucleic acid amplification method for pathogen detection that utilizes recombinase, single-strand binding protein (SSB), and DNA polymerase [5,6]. Combined with fluorescent probes or lateral flow strips, RPA enables rapid on-site detection of pathogens such as SARS-CoV-2 and Zika virus. However, its high sensitivity can sometimes lead to nonspecific amplification and compromise accuracy [5,6,7].
The CRISPR-Cas12 system, known for its high sensitivity and specificity, is a promising tool for genome editing, diagnostics, and pathogen detection [8,9]. RPA rapidly amplifies nucleic acids at constant temperatures, and CRISPR effectors (e.g., Cas12 or Cas13) confirm amplified products, amplify the signal, and reduce false positives. The combination of RPA and CRISPR enables faster, simpler, and more efficient assays, improving the specificity and sensitivity of nucleic acid detection in field and resource-limited settings, such as disease screening, food safety, and environmental monitoring [8,9,10]. However, maintaining RPA enzyme activity requires long-term storage at −20 °C, posing challenges for practical applications. Additionally, the RPA-CRISPR-Cas12a system involves multiple steps (nucleic acid extraction, RPA, and Cas12a binding), each requiring tube opening, which increases the risk of aerosol contamination and lengthens testing time [11,12].
To ensure accurate detection of aquatic pathogens, complex sample processing is required to obtain high-purity and high-concentration nucleic acids. Typical nucleic acid extraction methods for aquatic samples include centrifuge column-based [13] and magnetic bead-based [14] approaches. Some studies have also reported using direct thermal lysis to release nucleic acids, aiming to simplify sample processing [13]. However, these detection methods are intricate, time-consuming, and generally require well-trained personnel in specialized laboratories. Therefore, it is crucial to develop a new, simple, and rapid detection method for field detection of CAPRV2023, after RNA extraction, allows subsequent reverse transcription and amplification to be performed within a closed, one-step reaction system to meet urgent clinical diagnostic needs.
The present study introduces an innovative method based on a dual-signal amplification technique for nucleic acid detection of CAPRV2023 within a closed-tube system. The newly developed enzyme-mediated one-step sample processing–reverse transcription–enzyme-mediated duplex exponential amplification (EmOSP-RT-EmDEA) detection system integrates enzyme-mediated lysis and isothermal duplex exponential amplification into a single, closed-tube reaction. This system enables rapid, RNA extraction-free detection via a simplified one-step process. A concise overview of the reaction mechanism, including the role of RNA primers and the signal amplification strategy, is presented in Figure 1. This repetitive cycle effectively amplifies the detection signal, as illustrated in Figure 1.
This study presents an integrated CAPRV2023 EmOSP-RT-EmDEA detection method designed to streamline and enhance CAPRV2023 detection. The procedure begins with rapid RNA release from clinical samples of golden pompano using enzyme-mediated one-step sample processing (EmOSP) reagents. The resulting supernatant is directly introduced into the CAPRV2023 RT-EmDEA reaction, performed in a single closed-tube system at a constant temperature of 42 °C. Accurate results are obtained within 30 min, offering a powerful tool for monitoring and controlling CAPRV2023.
2. Materials and Methods
2.1. Clinical Sample Collection and RNA Preparation
Golden pompano exhibiting clinical symptoms of CAPRV2023 infection were collected from 37 geographically dispersed offshore cage farming sites across China between August 2023 and January 2025. A total of 113 clinical samples were obtained, with approximately three fish sampled per location. To minimize sampling bias, fish were randomly selected from each site regardless of size or sex. This sampling strategy was specifically designed to represent the natural variation in field conditions. The sampling period covered more than one complete production cycle of golden pompano, and the sampled fish represented all developmental stages, providing a comprehensive dataset across time, space, and developmental phases. RNA extraction was conducted following the protocol described by Sun et al. [1,2]. The CAPRV2023 strain was isolated from the affected golden pompano and preserved in the laboratory in 2023 [1,2].
2.2. Construction of Plasmid for Standard Quantification
A standard plasmid was constructed using CAPRV-740F/R primers (Table 1) following the protocol described by Sun et al. [1,2]. The plasmid concentration was measured at 136 ng/μL, corresponding to approximately 4.98 × 10^10^ copies/μL. It was then diluted to an appropriate concentration for use in subsequent experiments.
2.3. EmDEA Reaction System Development
2.3.1. Design of EmDEA Primers
Six forward primers, six reverse primers, and six RNA primers were designed based on the G protein gene sequence of CAPRV2023 (GenBank accession no. PP050495.1). The sequences are listed in Table 1.
2.3.2. Screening of EmDEA Reaction and Primers
The EmDEA reaction and primer screening were performed using the EmDEA Fluorescent Isothermal Amplification Screening Kit (GeneVide Biotech Co., Ltd., Suzhou, China), which contains enzyme dry powder and an activation solution. The components of the EmDEA Fluorescent Isothermal Amplification Screening Kit include: DNA amplification enzyme system (DNA recombinase, single-strand binding protein, T7 RNA polymerase, ATP regenerating enzyme, reverse transcriptase); signal amplification enzyme system (locating enzyme, cleavage enzyme, shrimp alkaline phosphatase, duplex-specific nuclease); and activation solution (nucleoside triphosphates, NTPs).
Each reaction mixture had a total volume of 20 μL, consisting of 1 μL of forward and reverse primers (10 μM each), 1 μL of RNA primer (100 μM), 7 μL of template DNA, enzyme dry powder, and 10 μL of activation solution.
A plasmid template containing 10^4^ copies was used for RNA probes screening, while decreasing concentrations of 10^3^ and 10^2^ copies were applied for forward and reverse primer screening, respectively. The reaction mixtures were incubated in a LightCycler^®^ 96 real-time quantitative PCR system (Roche, Basel, Switzerland) at a constant temperature of 42 °C, with fluorescence signals recorded every 60 s. The minimum threshold time (Tt) and maximum fluorescence intensity obtained were chosen as screening criteria. Primer combinations that performed well were further tested using a plasmid with only 10 copies, and only those demonstrating reliable amplification at this low copy number were selected for subsequent experiments.
2.4. Development of RT-EmDEA Reaction System
2.4.1. Preparation of RNA Standard Samples
RNA from tissues of CAPRV2023-infected golden pompano was quantified using a qPCR method developed by Sun et al. [1,2]. The Ct value was 15.74, corresponding to 10^7.11^ copies. This sample was diluted to the appropriate concentration for further use.
2.4.2. Reverse Transcription Primer Design and RT-EmDEA Reaction
To enable reverse transcription and the EmDEA reaction in a single closed tube, 20 short-sequence reverse transcription primers (Table 2) were designed and synthesized by Sangon Biotech Co., Ltd. (Shanghai, China) and prepared as 400 μM solutions. These primers were mixed equally to create an RT primer mix at a final concentration of 20 μM.
The RT-EmDEA reaction differs from the standard EmDEA reaction by incorporating reverse transcriptase into the enzyme dry powder. The final RT-EmDEA reaction mixture contained 1 μL of the RT primer mix, 1 μL each of forward and reverse primers (10 μM), 1 μL of RNA primer (100 μM), 6 μL of RNA template, and 10 μL of activation solution. The reaction mixture was incubated at 42 °C in a LightCycler^®^ 96 real-time qPCR system (Roche), with fluorescence readings collected every 60 s.
2.4.3. Screening of Compatible Primer Pairs for RT-EmDEA of CAPRV2023
The standard RNA sample prepared in Section 2.4.1 was diluted to 10^5^ copies as a template. Primer combinations selected in Section 2.3.2 were tested in the RT-EmDEA reaction following the methods described in Section 2.4.2 to assess compatibility. An effective primer pair generated a significant amplification curve, and the primer combination with the lowest Tt value was retained for subsequent steps.
2.4.4. Sensitivity of the CAPRV2023 RT-EmDEA Reaction
The standard RNA sample prepared in Section 2.4.1 was serially diluted to 5, 10, 10^2^, 10^3^, 10^4^, and 10^5^ copies and used as a template in the RT-EmDEA reaction to determine the system’s sensitivity.
2.5. Enzyme-Mediated One-Step Sample Processing Technique
2.5.1. Sample Processing Method
Three sample processing methods (Methods 1, 2, and 3) were developed to accommodate various experimental requirements. All methods utilized 1 mL of EmOSP lysis solution prepared in 1.5 mL Eppendorf tubes containing consistent components: 20 mM Tris-HCl (pH 8), 300 mM NaCl, 1 mM EDTA, 1% SDS, 0.8% PVP-40, and one engineered proteinase. The only difference among the three methods was the type of enzyme used: Method 1 used thermostable pronase, Method 2 used high-efficiency proteinase K, and Method 3 used thermostable papain. All reagents, including the enzymes, were purchased from GeneVide Biotech Co., Ltd., Suzhou, China. Samples were heated at 95 °C for 10 min, then cooled to room temperature, and the supernatant was collected for detection assays.
2.5.2. Minimally Invasive Sample Extraction
A 10 mg sample of gill filaments and feces from infected fish were collected and processed using each of the three methods described in Section 2.5.1. A 6 μL aliquot of the supernatant was then used in the RT-EmDEA reaction (described in Section 2.4.2) to assess the effectiveness of EmOSP. Each experiment was performed in triplicate. To ensure the reliability of sample extraction, the sample was also quantified according to the method described by Sun et al. [1,2].
2.5.3. Invasive Sample Extraction
Due to the high concentrations of CAPRV2023 in the spleen and liver, 10 mg of tissue samples from both tissues were collected and processed using the three methods described in Section 2.5.1. A 6 μL aliquot of the supernatant was analyzed using the RT-EmDEA reaction as outlined in Section 2.4.2. Each experiment was performed in triplicate, and quantification was confirmed following the method described by Sun et al. [1,2].
2.5.4. Environmental Sample Extraction
For environmental samples, 100 mL of seawater was collected from aquaculture cages where golden pompano were present and first filtered through a 0.22 μm pore-size membrane (Millipore, Burlington, MA, USA) to remove debris and particulates. The filtrate was then divided into two portions: one was directly used for EmOSP-based detection as described in Section 2.5.1, while the other was subjected to viral concentration using a 10 kDa molecular weight cut-off ultrafiltration tube (Millipore). The concentrated viral suspension was subsequently used for RNA extraction and RT-qPCR analysis. A 6 μL aliquot of the resulting supernatant was used for the RT-EmDEA reaction [1,2].
2.5.5. Optimization of EmOSP for Environmental Samples
Given that seawater salinity can influence EmOSP, the salinity of seawater was measured, and sterile seawater with matching salinity was prepared to dilute the test samples. CAPRV2023-containing seawater samples were diluted to various volumes and processed as described in Section 2.5.1. Optimization was assessed based on Tt and fluorescence values, with each test conducted in triplicate. Quantification was performed following the method outlined by Sun et al. [1,2].
2.6. EmOSP-RT-EmDEA Detection System Development for CAPRV2023
2.6.1. Integrating RT-EmDEA System for CAPRV2023
The EmDEA primer combination F2-R2-RNA4, along with the 20 RT primers and all RT-EmDEA enzyme components, were formulated into a single dry powder tube by GeneVide Biotech Co., Ltd. (China). For detection, only the test sample and activation solution are needed to initiate the reaction.
2.6.2. Specificity of the EmOSP-RT-EmDEA Detection System for CAPRV2023
RNA was extracted from viral pathogens, including VHSV, SCRV, SVCV, IHNV, NNV, and TiLV, and reverse-transcribed to cDNA. Additionally, DNA from ISKNV, SGIV, bacterial DNA from Streptococcus dysgalactiae, Vibrio alginolyticus, Streptococcus agalactiae, Streptococcus iniae, Lactococcus garvieae, Vibrio harveyi, and golden pompano DNA were tested. The cDNA from CAPRV2023-infected pompano was used as the positive control. Healthy fish cDNA and distilled water served as negative controls. Each experiment was performed in triplicate.
2.6.3. Repeatability of the EmOSP-RT-EmDEA Detection System for CAPRV2023
The system’s repeatability was assessed through inter- and intra-assay tests using the same sample. These tests were performed in triplicate across 10 independent runs in accordance with the protocols outlined in Section 2.5.3 and Section 2.4.3. The coefficient of variation (CV%) was calculated based on the mean Ct values.
2.6.4. Application of the CAPRV2023 EmOSP-RT-EmDEA Detection System
A total of 113 samples, including tissues, feces, and aquaculture water, were randomly collected from golden pompano aquaculture systems in Guangdong and Guangxi provinces between August 2023 and January 2025 for epidemiological surveillance and method comparison. These samples were tested using the CAPRV2023 EmOSP-RT-EmDEA Detection System, TaqMan qPCR, and conventional PCR methods [1,2]. Diagnostic sensitivity and specificity were determined as per OIE guidelines [14]. Viral infection was confirmed by observing typical cytopathic effects (CPEs) in FHM cells inoculated with the samples, which were used to verify CAPRV2023 infection status [1,2]. A Chi-square test was used to compare the detection rates of conventional PCR, TaqMan qPCR, and the CAPRV2023 EmOSP-RT-EmDEA system across the water, liver tissue, and feces samples. Results were coded as 1 for positive and 0 for negative. Statistical significance was determined at a significance level of 0.05, with p-values used to assess differences in the performance of the detection methods.
2.7. Clinical Samples and Ethics Statement
All fish were anesthetized using eugenol (clove oil) and humanely euthanized prior to dissection, in strict accordance with the approved animal ethics protocol. All animal experiments were conducted strictly following the recommendations in the ‘Guide for the Care and Use of Laboratory Animals’ set by the National Institutes of Health. All fish experiments were approved by the Animal Ethics Committee of Guangdong Provincial Key Laboratory of Aquatic Animal Disease Control and Healthy Culture, approval code: (20240621)006, approval date: 21 June 2024.
3. Results
3.1. Screening of RNA and EmDEA Primers
3.1.1. Screening of RNA Primer
Primer sets generating amplification signals with a Tt under 30 min were considered positive. Among these, the set with the lowest Tt was selected as the optimal primer for subsequent experiments due to its fastest amplification kinetics and highest reaction efficiency. Each combination was tested in triplicate to ensure reproducibility. Results of RNA primer screening are presented in Figure 2. Combinations involving RNA4 and RNA5 exhibited the shortest Tt and highest maximum fluorescence values among tested primers. Consequently, RNA4 and RNA5 were selected for further experiments based on these findings.
3.1.2. Screening of EmDEA Primer
The primer pair with the shortest Tt identified in Section 3.1.1 was further evaluated. Primer sets producing amplification signals with Tt below 30 min were considered positive. Using low Tt as the primary screening criterion, EmDEA was conducted with various primer pairs. As shown in Supplementary Table S1, six highly sensitive primer combinations were identified for further evaluation. Using a standard plasmid with 10 copies, four primer pairs—RNA4-F2-R2, RNA4-F2-R5, RNA5-F3-R2, and RNA5-F4-R2—were selected for subsequent experiments due to their ability to amplify at low copy numbers (Figure 3).
3.2. CAPRV-RT-EmDEA
3.2.1. RT-EmDEA Primer
Primer combinations selected in Section 3.1 were evaluated within the RT-EmDEA system using an RNA standard sample (prepared as described in Section 2.4.1) containing 10^4^ copies. As shown in Figure 4, only the F2-R2/F2-RNA4 combination produced a clear fluorescence amplification curve. F2-R5-RNA4 showed reduced sensitivity, while the other two pairs failed to amplify. Therefore, F2-R2-RNA4 was chosen as the optimal primer combination for the CAPRV2023-RT-EmDEA detection system.
3.2.2. Sensitivity of the CAPRV2023-RT-EmDEA
The sensitivity of the CAPRV2023-RT-EmDEA system was assessed using a serially diluted RNA standard sample. As shown in Figure 5, the detection limit was determined to be 4 copies/μL of RNA template under optimized reaction conditions, establishing this as the system’s detection threshold.
3.3. Enzyme-Mediated One-Step Sample Processing
3.3.1. Minimally Invasive Sample
RNA copy numbers in infected fish samples were quantified by TaqMan real-time PCR, and the extraction efficiency of minimally invasive samples processed using various EmOSP methods was evaluated with the CAPRV2023-RT-EmDEA system. In gill filament samples (10^4.83 ± 0.12^ copies/mg), none of the methods yielded sufficient nucleic acids for reliable amplification (Figure 6A). Conversely, in fecal samples (10^4.76 ± 0.11^ copies/mg), Methods 1 and 3 showed comparable extraction efficiencies, with Method 2 performing best (Figure 6B).
3.3.2. Invasive Sample
For spleen samples (10^6.03 ± 0.16^ copies/mg), RNA was successfully extracted in quantities sufficient for downstream detection (Figure 7B). For liver samples (10^6.07 ± 0.08^ copies/mg), Methods 1 and 3 demonstrated similar efficiencies, while Method 2 resulted in significantly longer Tt, indicating lower detection efficiency (Figure 7A).
3.3.3. Environmental Sample
RNA copy numbers in filtered seawater samples from CAPRV2023 disease outbreaks were quantified by TaqMan real-time PCR, and extraction efficiency of these samples processed with different EmOSP methods was assessed by the CAPRV2023-RT-EmDEA system. The seawater sample, with 33 ppt salinity and RNA concentration of 4.2 × 10^3^ copies/μL, showed similar extraction efficiencies across methods, with Method 2 performing the best (Figure 8).
To optimize seawater sample volume for EmOSP, CAPRV2023-containing seawater was serially diluted with sterile seawater (33 ppt salinity) for detection. As shown in Figure 9, 50 μL of seawater was optimal for lysis solution testing in the CAPRV2023-RT-EmDEA system.
The detection limit of the EmOSP-CAPRV2023-RT-EmDEA system was 4.2 × 10^1^ copies/μL under these optimized conditions for environmental samples (Figure 10).
3.4. CAPRV2023 EmOSP-RT-EmDEA Detection System
3.4.1. Specificity of the CAPRV2023 EmOSP-RT-EmDEA Detection System
Specificity was evaluated using a panel of common golden pompano pathogens and several viruses phylogenetically related to CAPRV2023 to ensure clinical and molecular specificity. As shown in Figure 11, amplification curves appeared exclusively for CAPRV2023; no amplification was detected for any other pathogens.
3.4.2. Repeatability of the CAPRV2023 EmOSP-RT-EmDEA Detection System
Both intra-assay repeatability and inter-assay reproducibility of the CAPRV2023 EmOSP-RT-EmDEA detection system were evaluated by calculating the CV of Tt values, using clinical liver samples containing (10^7.59 ± 0.12^ copies/mg) of viral RNA. Ten independent reaction groups were tested, each performed in triplicate. As shown in Figure 12, the Tt values across inter- and intra-groups exhibited good consistency with minimal variation. The CVs for intra-assay ranged from 3.29% to 6.82%, with standard deviations of 0.13 to 0.58 (Figure 13). The CV for inter-assay was 2.51% (Figure 13).
3.4.3. Application
To comprehensively assess the distribution and detectability of CAPRV2023 in golden pompano under real-world aquaculture conditions, 113 clinical and environmental samples were collected from 37 geographically distinct aquaculture sites across China. These sites spanned more than one production cycle and included golden pompano of all developmental stages (Figure 14). All samples were tested using three molecular assays: conventional PCR, TaqMan qPCR, and the EmOSP-RT-EmDEA system.
As summarized in Supplementary Table S2, the EmOSP-RT-EmDEA system demonstrated a markedly higher positive detection rate in seawater samples (76%) compared to conventional PCR (16%) and was nearly equivalent to TaqMan qPCR (79%). Statistical analysis using Fisher’s exact test confirmed no significant difference in seawater detection rates between EmOSP-RT-EmDEA and TaqMan qPCR (p = 0.7351), while both methods significantly outperformed conventional PCR (p < 0.0001). Detailed comparative data are provided in Supplementary Table S2 for clarity.
A similar trend was observed in spleen and fecal samples. The EmOSP-RT-EmDEA system achieved positive rates of 69.03% and 68.14%, respectively, consistent with TaqMan qPCR results. Fisher’s exact test revealed no significant differences between EmOSP-RT-EmDEA and TaqMan qPCR for these samples (p = 1.000 for both spleen and fecal samples), while both showed significantly higher sensitivity than conventional PCR (p = 0.0001 for spleen; p < 0.0001 for feces).
Collectively, these results demonstrate that the EmOSP-RT-EmDEA detection system provides field-applicable sensitivity comparable to TaqMan qPCR across environmental and clinical matrices, making it a promising alternative for rapid CAPRV2023 surveillance in aquaculture environments.
4. Discussion
A recent outbreak of a devastating viral disease caused by CAPRV2023 has severely impacted net cage-cultured golden pompano in China, posing a significant threat to the aquaculture industry. As reported by Sun et al. [1,2], golden pompano infected with CAPRV2023 exhibit high mortality rates within 7 to 10 days. Currently, no effective prevention or control measures exist for this epidemic. Therefore, early detection of this emerging virus during initial infection stages, coupled with timely intervention, is crucial for sustainable development of golden pompano aquaculture. In this study, a novel nucleic acid assay for CAPRV2023 based on a dual-signal amplification technique was developed and tested in the field. The assay demonstrated strong specificity, high sensitivity, excellent repeatability, and stability.
The G protein of fish rhabdoviruses is essential for membrane fusion and viral virulence and acts as a key antigen inducing virus-neutralizing antibodies and host immunity [14,15,16,17,18,19]. Given its high transcription levels in infected hosts, the G protein gene is an ideal molecular detection target, previously applied for IHNV and VHSV detection [20,21]. In this study, the CAPRV2023 EmOSP-RT-EmDEA detection system targeting the G protein gene showed high specificity and sensitivity, detecting the virus in infected fish without cross-amplification of other fish pathogens.
To address limitations of existing CAPRV2023 diagnostics, we evaluated conventional PCR, TaqMan qPCR, and CRISPR-Cas platforms. Conventional PCR and TaqMan qPCR are widely used, their multi-step workflows require RNA extraction, reverse transcription, and amplification in open-tube formats. These procedures increase aerosol contamination risk and involve time-consuming, instrument-dependent operations [22,23]. Dye-based qPCR detects total amplified product via fluorescence intensity, whereas probe-based qPCR uses hydrolysis probes [24]. Both rely solely on product accumulation for signal generation. Although TaqMan qPCR offers high sensitivity (2 copies/μL) and strong quantification, it requires precise thermal cycling equipment, enzyme storage at −20 °C, and at least three processing steps (extraction, reverse transcription, and amplification), limiting field applicability (Figure 15). The RPA–Cas12a detection approach can be categorized into two formats. The first is a two-tube method, in which RPA is performed first, followed by opening the tube to add the Cas12a reaction mixture. The second is a one-tube method, which eliminates the need for tube opening but requires a substantially higher concentration of Cas12a protein, resulting in a significantly increased cost (Figure 16). Detection of environmental samples using conventional methods is even more complex. For instance, TaqMan qPCR requires raw seawater to undergo filtration (0.22 μm pore size), ultrafiltration concentration, RNA extraction, reverse transcription, and amplification—five separate steps requiring specialized equipment. These additional steps increase cost, cross-contamination risk, and total turnaround time.
In contrast, RPA enables rapid isothermal nucleic acid amplification at constant low temperatures (typically around 37–42 °C) [25]. RPA eliminates the need for thermal cyclers and significantly reduces reaction time [25]. However, RPA is known to exhibit a relatively high false-positive rate due to nonspecific primer interactions under isothermal conditions, especially in complex sample matrices [26]. To improve specificity, many RPA-based methods still rely on open-tube formats for downstream detection and incorporate CRISPR-Cas enzymes for post-amplification detection [27,28,29]. These CRISPR-Cas systems enhance signal fidelity through collateral cleavage mechanisms but require multi-step workflows [27,28,29]. Reopening the reaction tube after RPA to introduce the CRISPR-Cas detection system poses a substantial contamination risk, as the reaction contains high-copy, low-molecular-weight amplicons that are highly susceptible to aerosolization and environmental dispersion [10,11,29,30,31]. Furthermore, these systems depend on temperature-sensitive reagents and specialized components, such as purified Cas proteins and guide RNAs, limiting their practicality in field settings [32,33,34].
To overcome these limitations, we developed the CAPRV2023 EmOSP-RT-EmDEA detection system, which integrates enzyme-mediated one-step sample processing, reverse transcription, and duplex exponential isothermal amplification into a fully enclosed, two-step workflow. This design eliminates the requirement for RNA purification or centrifugation, thereby minimizing contamination risk by avoiding tube reopening. It enables direct input of raw biological or environmental samples through their addition to lysis buffer tubes, where a simplified enzyme-mediated lysis step is performed. The resulting lysate can be directly used in downstream amplification without requiring conventional magnetic bead or column-based nucleic acid extraction, simplifying the workflow and reducing equipment needs (Figure 1).
As illustrated in Figure 15, the EmOSP-RT-EmDEA system delivers results in approximately 30 min with only two handling steps. It operates without thermal cyclers and supports reagent storage at 4 °C or room temperature, reducing equipment and cold-chain requirements. Compared to conventional protocols—which typically involve 3–5 discrete operations including extraction, reverse transcription, and amplification—the EmOSP approach dramatically streamlines the workflow and reduces the need for highly trained personnel. The EmOSP-RT-EmDEA system incorporates a dual-signal amplification mechanism, wherein cDNA is transcribed into RNA to exponentially enhance detection sensitivity. This strategy enables identification of low-abundance viral targets, minimizing false negatives and supporting robust field testing. The system achieved a detection limit of 4 copies/μL, closely approaching TaqMan qPCR (2 copies/μL), exceeding SYBR Green I qPCR by over 20-fold [1,2], and vastly outperforming conventional PCR (1000 copies/μL). These sensitivity advantages, along with excellent intra-assay consistency, considerably enhance the assay’s applicability for rapid, on-site diagnosis in aquaculture environments.
The assay was rigorously validated across various biological and environmental matrices. Gill tissues were found to be unsuitable for effective nucleic acid release due to low nucleic acid yield, whereas feces proved to be a minimally invasive and reliable sampling alternative. As part of a large-scale surveillance initiative, 113 clinical samples representing all life stages of golden pompano were collected from 37 aquaculture sites along the South China Sea, covering more than one complete production cycle (Figure 14). This broad geographic and temporal sampling demonstrated the robustness and field applicability of the EmOSP-RT-EmDEA system. As summarized in Supplementary Table S2, the system achieved positive detection rates of 76.0% in seawater, 69.03% in spleen tissue, and 68.14% in feces, closely matching the TaqMan qPCR results (79.0%, 69.03%, and 68.14%, respectively), and significantly outperforming conventional PCR (16.0%, 40.71%, and 32.7%, respectively). These results highlight the reliability and diagnostic effectiveness of the EmOSP-RT-EmDEA system under real-world aquaculture conditions.
In environmental testing, the EmOSP-RT-EmDEA system achieved a 76.0% positive detection rate in raw seawater samples without any pre-concentration steps. Although TaqMan qPCR showed slightly higher detection rates (79.0%), this advantage can be attributed to its use of ultrafiltration-based viral concentration prior to amplification [35]. Importantly, the EmOSP-RT-EmDEA system demonstrated the capacity to detect as few as 4.2 copies per sample directly, underscoring its sensitivity and suitability for on-site aquaculture monitoring. This capability aligns well with epidemiological needs, as prior studies have shown that CAPRV2023 infection can be initiated via waterborne exposure [1,2]. Therefore, effective detection of free virus in seawater—without complex sample processing—provides a practical early-warning approach for disease control in aquaculture operations.
In summary, the EmOSP-RT-EmDEA system offers a closed-tube, rapid, and highly sensitive nucleic acid detection platform that simplifies sample processing, minimizes contamination risk, and requires minimal equipment. These advantages establish it as an effective diagnostic and monitoring tool for CAPRV2023 in both clinical and environmental aquaculture settings. Moreover, the EmOSP-RT-EmDEA system provides a reliable and field-deployable molecular approach for monitoring viral pathogens in aquaculture and shows great potential for seafood hygiene surveillance and raw-material quality control throughout the aquatic food production chain.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sun H. Fred B. Wang H. Huang J. Wu Z. Wu D. Lu Y. Jian J. Huang Y. One-step and two-step q PCR assays for CAPRV 2023: Development and application in full-cycle epidemiological surveillance of golden pompano Front. Vet. Sci.202512162099710.3389/fvets.2025.162099740822660 PMC 12351644 · doi ↗ · pubmed ↗
- 2Sun H. Huang J. Wang H. Zhang Y. Fei Q. Zhou J. Yang L. Li Y. Jian J. Lu Y. Mass mortality associated with Carpione rhabdovirus in golden pompano (Trachinotus ovatus) in China: First report Aquaculture 202559574163810.1016/j.aquaculture.2024.741638 · doi ↗
- 3PRC 2024 China Fishery Products Report FAS China Staff PRC Beijing, China 2024
- 4Bovo G. Olesen N. Jørgensen P. Ahne W. Winton J. Characterization of a rhabdovirus isolated from carpione Salmo trutta carpio in Italy Dis. Aquat. Org.19952111512210.3354/dao 021115 · doi ↗
- 5Daher R.K. Stewart G. Boissinot M. Bergeron M.G. Recombinase Polymerase Amplification for Diagnostic Applications Clin. Chem.20166294795810.1373/clinchem.2015.24582927160000 PMC 7108464 · doi ↗ · pubmed ↗
- 6Balea R. Pollak N.M. Hobson-Peters J. Macdonald J. Mc Millan D.J. Development and pre-clinical evaluation of a Zika virus diagnostic for low resource settings Front. Microbiol.202314121414810.3389/fmicb.2023.121414838053551 PMC 10694267 · doi ↗ · pubmed ↗
- 7Zyrina N.V. Antipova V.N. Nonspecific Synthesis in the Reactions of Isothermal Nucleic Acid Amplification Biochemistry 2021868878973428471310.1134/S 0006297921070099 · doi ↗ · pubmed ↗
- 8Chen J.S. Ma E. Harrington L.B. Da Costa M. Tian X. Palefsky J.M. Doudna J.A. CRISPR-Cas 12a target binding unleashes indiscriminate single-stranded D Nase activity Science 201836043643910.1126/science.aar 624529449511 PMC 6628903 · doi ↗ · pubmed ↗