Prenatal Diagnosis of 6q Terminal Deletion Associated with Coffin–Siris Syndrome: Phenotypic Delineation and Review
Christian Peña-Padilla, David Alejandro Martínez-Ceccopieri, Evelin Montserrat García-Hernández, Lucina Bobadilla-Morales, Jorge Román Corona-Rivera

TL;DR
This paper reports a prenatal diagnosis of a 6q terminal deletion linked to Coffin–Siris Syndrome and highlights key clinical features associated with this rare genetic condition.
Contribution
The study provides a detailed phenotypic delineation of 6q25.3 deletions involving the ARID1B gene and their association with Coffin–Siris Syndrome.
Findings
Facial gestalt, hypertrichosis, and fifth fingernail aplasia/hypoplasia are key features of CSS linked to 6q25.3 deletions.
Vertebral defects and cystic hygroma are additional clinical signs associated with ARID1B gene deletions.
Prenatal diagnosis of 6q terminal deletion can lead to early identification of Coffin–Siris Syndrome.
Abstract
Chromosome 6q deletion syndrome is a rare entity that has a highly variable clinical presentation and size of deletions. The most frequent manifestations of 6q terminal deletion are intellectual disability, facial dysmorphism, brain structural anomalies, and congenital heart defects. The phenotype is not clinically recognizable, except in those who harbor a terminal 6q deletion that includes the ARID1B gene, in whom features similar to Coffin–Siris syndrome (CSS) can be observed. We report the case of a female newborn with a prenatal diagnosis of a terminal deletion on 6q25.1q27, which encompasses the ARID1B gene, and who was diagnosed with CSS during the neonatal period. From our review, we found that facial gestalt, hypertrichosis, and fifth fingernail aplasia/hypoplasia, along with other features, such as vertebral defects and cystic hygroma (or webbed neck), correlated with the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
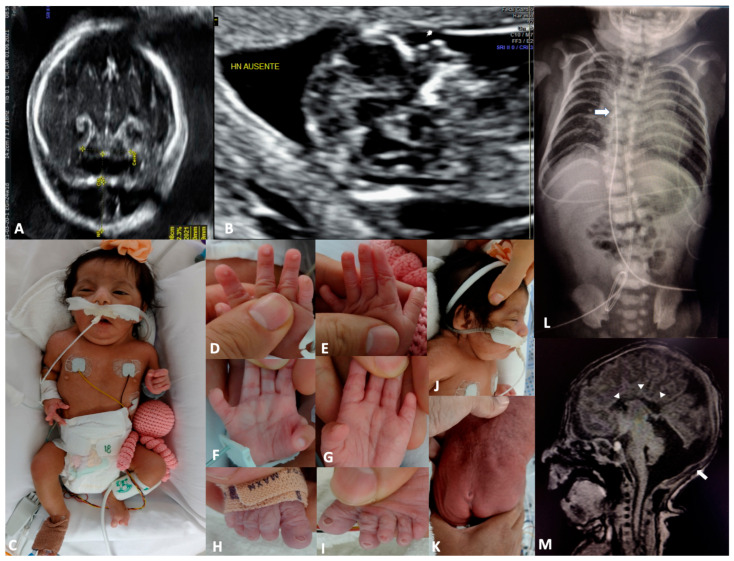 Figure 1
Figure 1 Figure 2
Figure 2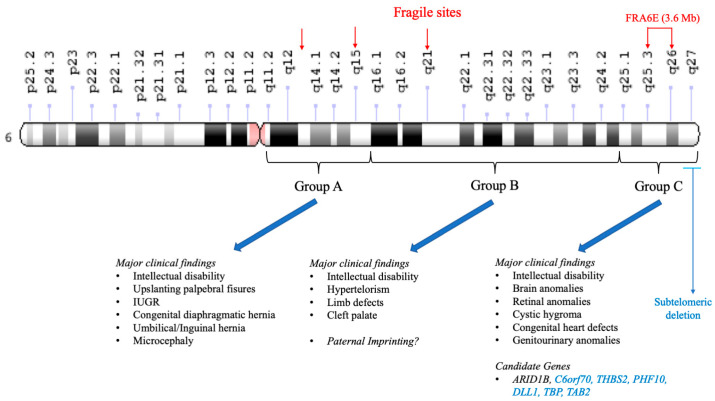 Figure 3
Figure 3| Reference | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [ | [ | [ | [ | [ | [ | [ | [ | [ | [ | [ | [ | [ | Present Case | ||
| Patient | 2 | 1 | 2 | 4 | 3 | 5 | 4 | 4 | |||||||
| Sex | M | M | M | M | M | F | M | F | M | M | F | F | M | F | F |
| Age at last examination | 2 yr | 9 mo | 3 yr | 1 yr | 21 wk | Fetus | 4 mo | 2 yr | 10 yr | 2 yr | 37 yr | 9 yr | 10 mo | 18 mo | 3 mo |
|
| |||||||||||||||
| Intrauterine growth retardation | − | − | − | − | + | − | − | − | − | − | NS | − | − | + | − |
| Cystic hygroma | − | − | − | − | + | + | − | − | − | − | NS | − | − | − | + |
| Oligohydramnios | − | − | − | − | − | − | − | − | − | − | NS | − | − | + | − |
| Ventriculomegaly | − | − | − | − | + | + | − | − | − | − | NS | − | + | − | − |
| Hydrops fetalis | − | − | − | − | + | − | − | − | − | − | NS | − | − | − | − |
| Hydrothorax | − | − | − | − | − | − | − | − | − | − | NS | − | − | − | − |
| Absent nasal bone | − | − | − | − | − | − | − | − | − | − | NS | − | − | − | + |
| Diaphragmatic hernia | − | − | − | − | + | − | − | − | − | − | NS | − | − | − | − |
|
| |||||||||||||||
| Birth length cm (percentile) | NS | NS | 51 (P50) | 48 (P60) | 23 (P25) | NS | 44 | NS | NS | 55.5 (P5) | NS | 49 (P50) | 47 (P1) | 48 | 41 (P10) |
| Birth weight g (percentile) | NS | 2670 (P1) | 3600 (P54) | 2870 (P60) | 280 (P10) | NS | 2220 | 1800 | NS | 2600 (P10) | NS | 2800 (P25) | 2800 (P3) | 2000 | 1840 (P16) |
| OFC at birth cm (percentile) | NS | NS | 35.5 (P50) | 32 (<P3) | 16 (P25) | NS | 30.8 | NS | NS | 44 (>P95) | NS | 34 (P50) | 31 (<P1) | 30 | 32 (P76) |
| Developmental delay/intellectual disability | + | + | + | + | NA | NA | + | + | + | + | + | + | + | + | + |
| Microcephaly | Yes | + | + | + | − | NS | + | − | − | − | NS | − | + | + | − |
| Sparse scalp hair | − | + | − | − | NS | − | + | − | − | + | − | − | − | − | − |
| Thick eyebrows | − | − | − | − | NS | − | − | − | − | − | + | − | + | − | + |
| Long eyelashes | − | − | − | − | NS | − | − | − | − | − | NS | − | + | − | + |
| Dacryostenosis | − | − | − | NS | NS | − | − | − | NS | + | NS | − | − | − | − |
| Flat nasal bridge | − | + | + | − | + | − | + | + | − | − | − | + | − | + | + |
| Thick alae nasi | − | + | + | − | − | NS | − | − | − | − | + | − | − | − | + |
| Anteverted nose | − | + | − | + | + | + | + | − | − | + | − | − | − | + | + |
| Long philtrum | + | + | + | + | + | NS | + | + | − | + | − | − | + | − | + |
| Large mouth | + | − | + | + | − | + | − | + | + | − | + | − | − | − | + |
| Dysplastic ears | + | − | + | − | + | + | + | + | − | + | + | + | + | + | + |
| Hypertrichosis | − | − | − | + | − | − | − | + | − | − | NS | − | NS | − | + |
| Fifth fingernail aplasia/hypoplasia | − | − | − | + | − | NS | − | + * | − | NS | NS | − | − | − | + |
| Brachydactyly | + | − | − | − | − | NS | − | NS | No | NS | NS | − | − | + | + |
| Hemivertebrae | NS | NS | NS | NS | − | NS | NS | NS | NS | NS | NS | − | NS | NS | + |
|
| |||||||||||||||
| Atrioventricular septal defect | − | − | − | − | + | − | − | + | − | + | NS | − | − | − | − |
| Ventricular septal defect | − | + | − | − | − | + | + | − | − | − | NS | − | − | + | + |
| Atrial septal defect | − | − | − | − | − | − | − | − | − | − | NS | − | + | + | + |
| Partially anomalous pulmonary venous drainage | − | − | + | − | − | − | − | − | − | − | NS | − | − | − | − |
|
| − | − | − | − | − | − | − | + | − | − | NS | − | − | − | − |
| Pulmonary vein stenosis | − | − | − | − | − | − | − | − | − | + | NS | − | − | − | − |
|
| |||||||||||||||
| Hydronephrosis | + | − | − | − | − | + | − | − | − | + | − | − | − | − | − |
| Duplicated collecting system | − | − | − | − | + | − | − | − | − | − | − | − | − | − | − |
| Renal cyst | + | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Cryptorchidism | + | + | − | − | − | NA | + | NA | − | − | NA | NA | − | NA | NA |
| Penoscrotal webbing | − | − | − | − | − | − | − | − | − | − | − | − | + | − | − |
| Clitoromegaly | − | − | − | − | − | − | − | − | − | − | − | − | − | + | − |
| Prominent labia minora | − | − | − | − | − | − | − | − | − | − | − | − | − | + | − |
|
| |||||||||||||||
| Corpus callosum agenesis (A)/hypoplasia | NS | NS | NS | −/− | +/− | −/− | +/− | −/− | NS | −/− | −/+ | −/+ | +/− | +/− | +/− |
| Ventriculomegaly/hydrocephaly | NS | NS | NS | −/− | −/− | −/− | −/− | +/− | NS | −/+ | −/− | −/− | −/− | −/− | −/− |
| Colpocephaly | NS | NS | NS | − | − | − | − | − | NS | − | + | + | + | − | − |
| Arhinencephaly | NS | NS | NS | − | + | − | − | − | NS | − | − | − | − | − | − |
| Cerebellar hypoplasia | NS | NS | NS | − | − | − | − | − | NS | − | − | − | − | − | + |
| Other anomalies | HD, PrH | CP, HD | Cd | PrH | CP, DH | DH, SUA | AA, CP, PrT, STTh | TGC, RNS | − | EPi, RPi | S | − | S | − | RNS |
|
| |||||||||||||||
| Cytogenetic band deleted | 6q25 | 6q25 | 6q24 | 6q25 | 6q23 | 6q24.3 | 6q24.3 | 6q25.3 | 6q25 | 6q25.2 | 6q25.3 | 6q25.2 | 6q25.3 | 6q24 | 6q25.1 |
| ? | ? | + | + | + | + | + | ? | ? | + | + | + | + | + | + | |
| CSS clinically recognizable | − | − | − | + | − | − | − | + | − | − | − | − | − | − | + |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsChromatin Remodeling and Cancer · Genomic variations and chromosomal abnormalities · Melanoma and MAPK Pathways
1. Introduction
Chromosome 6q deletion syndrome (C6qDS) is a rare entity first described by Milosevic and Kalicanin in 1975 [1] in a boy with dysmorphic features and developmental delay. Since then, several cases have been reported. The clinical recognition of C6qDS is challenging due to the variation in deletion size and genes involved. In 1997, Hopkin et al. proposed a classification of C6qDS into three groups based on the description of 60 reported cases: (A) proximal deletion (6q11-q16), (B) middle deletion (6q15-q25), and (C) terminal deletion (6q25-qter) [2]. Intellectual disability is present in all groups. Group A correlates with mild dysmorphic features and lower frequency of congenital heart defects (CHDs) and microcephaly; Group B correlates with limb anomalies and high neonatal mortality; and Group C correlates with a higher incidence of brain anomalies, but remains difficult to clinically recognize. Advances in molecular cytogenetics have broadened the clinical spectrum, identifying microdeletions and new candidate genes [3,4,5,6,7,8,9].
On the other hand, Coffin–Siris syndrome (CSS) was first described by Coffin and Siris in 1970 in three unrelated females with intellectual disability, coarse facial features, and absence of the fifth fingernail [10]. Not until 2012 did Santen et al. identify pathogenic variants in the ARID1B gene as the etiology of CSS type 1 (CSS1) [11], and in the same year, Tsurusaki et al. reported other CSS genes related to the SWI/SNF complex [12]. The ARID1B gene (6q25.3) was proposed as a candidate gene for cerebral dysgenesis in terminal 6q deletion syndrome by Backx et al. 2011 [13] and later by Michelson et al. 2012 in a boy with an interstitial deletion at 6q25, including only the genes ARID1B and ZDHHC [14]. Most patients with CSS do not have prenatal US anomalies, and those reported are mainly congenital diaphragmatic hernia (CDH), congenital heart defects (CHDs), and intrauterine growth restriction (IUGR). However, in CSS related to ARID1B, the most frequent anomalies detected by prenatal US are brain anomalies, including corpus callosum agenesis (CCA) [15]. Here, we report a case of a female newborn with a prenatally detected 6q terminal deletion, which encompasses the ARID1B gene, in whom classic CSS was clinically diagnosed at birth. Additionally, we review all previous reports with similar deletions on chromosome 6q.25.3.
2. Materials and Methods
Written informed consent was obtained from the parents for publication. Amniotic fluid was collected via transabdominal amniocentesis. Fetal cells were cultured in situ and prepared for G-banded karyotyping. Chromosomal analysis was performed on metaphase spreads, with 20 cells counted. The karyotype was interpreted and reported according to ISCN, 2020. Genomic DNA was extracted from peripheral blood leukocytes using a commercial extraction kit, according to the manufacturer’s protocol. aCGH was performed using the commercial oligonucleotide microarray platform KaryoNIM^®^60K. Data were analyzed using Agilent Cytogenomics Agilent Technologies, Inc., Santa Clara, California, USA Software v5.0.2.5 [16]. Detected copy number variations (CNVs) were filtered and classified following the guidelines of ACMG.
3. Case Report
The proposita was the first child of a young, healthy, and non-consanguineous couple. No family history of miscarriages or malformations was reported. During the first trimester, a fetal ultrasound detected a cystic hygroma (CH) and absence of the nasal bone (Figure 1A,B). At 18 weeks of gestation, amniocentesis and karyotyping were performed, reporting 46, XX, del(6)(q23) (Figure 2A). Despite prenatal diagnosis, the parents decided to continue the pregnancy. She was born at the 34th week of gestation by cesarean section. The Apgar score was 7–8 at 3 and 5 min, respectively. At birth, weight was 1840 g (P16), height was 41 cm (P10), and OFC was 32 cm (P76). Physical examination revealed coarse facial features with generalized hypertrichosis, dysplastic ears, a short neck with redundant nuchal skin, a broad thorax with widely spaced nipples, short hands with brachydactyly, nail hypoplasia of fingers 1–4, and nail aplasia of the left fifth finger. The feet showed nail aplasia of the fifth toes. A deep sacral skin dimple was also noted (Figure 1C–K). Thorax radiography revealed a hemivertebra in T6 (Figure 1L). Brain MRI showed agenesis of the corpus callosum, cerebellar hypoplasia, and pseudocystic dilatation of the posterior fossa (Figure 1M). Echocardiogram showed an ostium secundum atrial septal defect (ASD), along with a ventricular septal defect (VSD). Renal ultrasonography was normal. A CGH array was performed, revealing arr [16] 6q25.1q27 (150321670_170537245)x1 (Figure 2B). The deletion spanned 20.2 megabases, involving at least 74 genes, spanning morbid genes as SYNE1, ARID1B, PDE10A, DLL1, and TBP (Figure 2B, lower panel in the enlargement). A normal karyotype was obtained for both parents. The proposita died at three months of age due to sepsis and multiorgan failure; an autopsy was not performed.
4. Discussion
Pure chromosome 6q terminal deletions are relatively rare, with a frequency of about 0.05% in patients with intellectual disability and multiple malformations [9]. Although deletions across all chromosome 6q have been described, most recent research has focused on subtelomeric deletions in the cytoband 6q27, adding a pure subtelomeric deletion subgroup for the original Group C proposed by Hopkin et al. [2]. Regarding this subgroup, some candidate genes (DLL1, THBS2, PHF10, and C6orf70) have been proposed to explain the observed brain anomalies [4,5,6,8,9,17]. Expanding this subject, in 2005, Eash et al. described a correlation between deletion spans 6q26-q27 with congenital heart defects, genital hypoplasia, a short neck, and retinal anomalies, and pure subtelomeric deletions with dysmorphic features and brain anomalies, including ventriculomegaly/hydrocephaly [18]. Our patient, along with the 14 other cases summarized in Table 1, was analyzed to establish possible specific genotype–phenotype correlations, as detailed below.
A key objective of our analysis was to correlate specific genetic loci within Group C with distinct phenotypes. We noted that ARID1B gene haploinsufficiency (proximal at Group C, 6q25.1) correlates with the distinctive nail hypoplasia/aplasia of the fifth finger. This recognizable clinical manifestation is observed in 68% of patients with CSS1 [19]. Our patient is one of only three terminal 6q deletion cases to date that exhibit a recognizable CSS phenotype (along with Stevens et al. [20] and Meng et al. [21]). This finding suggests that the CSS phenotype in other 6q terminal deletion cases may be masked by haploinsufficiency of other contiguous subtelomeric genes. This is supported by the fact that only patients with small interstitial deletions, including ARID1B, display a more evident CSS phenotype [14,19,22]. Other seemingly specific features were hypertrichosis and corpus callosum agenesis (Table 1). Our case is particularly significant due to the presence of cystic hygroma (CH), a feature previously reported only in a few patients harboring deletions between 6q23 and 6q27 [23,24,25]. Also, Meng et al. reported a case with a webbed neck and karyotype 46, XX, del(6)(q25) [21], suggesting that CH is a clinical feature in terminal deletion but not in subtelomeric deletion only. No genes in this chromosomal region are known to be involved in CH or lymphatic system development. All five patients had CHD, specifically AVD and VSD, except for those reported by Shen-Shwartz et al. and Meng et al. [21,23], in which an atrioventricular septal defect was reported, leading to the suspicion of a relationship between CHD and CH in C6qDS patients.
The clinical variability among case reports could be explained by the wide range of deletion sizes. Chromosome 6q contains several known fragile sites (q13, q21, and q26) [26], in addition to a preferential breakpoint at 6q25 first noted by Valtat et al. in 1992 [25]. This region was later characterized as FRA6E, a fragile site spanning 3.6 Mb from 6q25.3 to 6q26 [27] (Figure 3), resulting in a highly variable deletion size and, consequently, diverse clinical outcomes.
5. Conclusions
Chromosome 6q deletion syndrome is a rare chromosomal abnormality with a highly variable clinical presentation, making it very difficult to clinically recognize. The typical features of CSS, along with other anomalies, such as vertebral defects and CH (webbed neck), appear to correlate with the presence of CSS related to small deletions on 6q25.3 that include the ARID1B gene. Further large cohort studies are required to better understand its clinical variability and identify potential interactions between genes surrounding ARID1B.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1MilosevićJ. Kalicanin P. Long arm deletion of chromosome no. 6 in a mentally retarded boy with multiple physical malformations J. Ment. Defic. Res.19751913914410.1111/j.1365-2788.1975.tb 01266.x 1195357 · doi ↗ · pubmed ↗
- 2Hopkin R.J. Schorry E. Bofinger M. Milatovich A. Stern H.J. Jayne C. Saal H.M. New insights into the phenotypes of 6q deletions Am. J. Med. Genet.19977037738610.1002/(SICI)1096-8628(19970627)70:4<377::AID-AJMG 9>3.0.CO;2-Q 9182778 · doi ↗ · pubmed ↗
- 3Caselli R. Mencarelli M. Papa F. Uliana V. Schiavone S. Strambi M. Pescucci C. Ariani F. Rossi V. Longo I. A 2.6 Mb deletion of 6q 24.3-25.1 in a patient with growth failure, cardiac septal defect, thin upperlip and asymmetric dysmorphic ears Eur. J. Med. Genet.20075031532110.1016/j.ejmg.2007.03.00317512813 · doi ↗ · pubmed ↗
- 4Mosca A. Callier P. Masurel-Paulet A. Thauvin-Robinet C. Marle N. Nouchy M. Huet F. Dipanda D. De Paepe A. Coucke P. Cytogenetic and array-CGH characterization of a 6q 27 deletion in a patient with developmental delay and features of Ehlers-Danlos syndrome Am. J. Med. Genet. A 2010152 A 1314131710.1002/ajmg.a.3325420425843 · doi ↗ · pubmed ↗
- 5Gerber J.C. Neuhann T.M. Tyshchenko N. Smitka M. Hackmann K. Expanding the clinical and neuroradiological phenotype of 6q 27 microdeletion: Olfactory bulb aplasia and anosmia Am. J. Med. Genet. A 20111551981198610.1002/ajmg.a.3407921744487 · doi ↗ · pubmed ↗
- 6Rigon C. Salviati L. Mandarano R. DonàM. Clementi M. 6q 27 subtelomeric deletions: Is there a specific phenotype?Am. J. Med. Genet. A 20111551213121410.1002/ajmg.a.3387721484997 · doi ↗ · pubmed ↗
- 7Nowaczyk M.J. Carter M.T. Xu J. Huggins M. Raca G. Das S. Martin C.L. Schwartz S. Rosenfield R. Waggoner D.J. Paternal deletion 6q 24.3: A new congenital anomaly syndrome associated with intrauterine growth failure, early developmental delay and characteristic facial appearance Am. J. Med. Genet. A 2008146 A 35436010.1002/ajmg.a.3214418203180 · doi ↗ · pubmed ↗
- 8Peddibhotla S. Nagamani S.C.S. Erez A. Hunter J.V. Holder J.L.Jr. Carlin M.E. Bader P.I. Perras H.M.F. Allanson J.E. Newman L. Delineation of candidate genes responsible for structural brain abnormalities in patients with terminal deletions of chromosome 6q 27Eur. J. Hum. Genet.201523546010.1038/ejhg.2014.5124736736 PMC 4266737 · doi ↗ · pubmed ↗