The microbiome’s hidden influence: preclinical insights into inflammatory responses in necrotizing enterocolitis
Briana M. Peterson, Ina Rudloff, Nadia S. Deen, Sara K. Di Simone, Ramesh M. Nataraja, Gergely Toldi, Maurizio Pacilli, Steven P. Garrick, Steven X. Cho, Marcel F. Nold, Samuel C. Forster, Claudia A. Nold-Petry

TL;DR
This paper reviews how the microbiome and immune system interact in early life, focusing on their role in necrotizing enterocolitis and potential therapeutic strategies.
Contribution
The paper provides a comprehensive review of preclinical insights into microbiome-immune interactions in NEC, highlighting gaps and opportunities for new therapies.
Findings
Prematurity, dysbiosis, and underdeveloped immunity are key risk factors for NEC.
Animal models and bacterial genomic analyses reveal the microbiome's critical role in NEC pathophysiology.
Probiotics show moderate preventive effects, supporting microbiome-targeted interventions.
Abstract
Necrotizing enterocolitis (NEC) is the most common surgical emergency in preterm infants; nonetheless, besides supportive measures, no treatment is available. NEC significantly increases length of hospitalization of preterm infants, causes severe morbidity and up to 70% mortality. Despite limited understanding of the underlying mechanisms, prematurity, dysbiosis and an underdeveloped immune system are known to increase the risks of developing NEC. The low weight of preterm infants (often < 2000 g) and unpredictable progression of NEC hinder clinical research; hence, most of our mechanistic understanding of NEC pathophysiology has arisen from animal models. Recent advances in bacterial genomic analyses highlighted the intestinal microbiome’s key role in NEC, strengthening the concept that this disease results from an interaction between the patient’s developing immune system and their…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
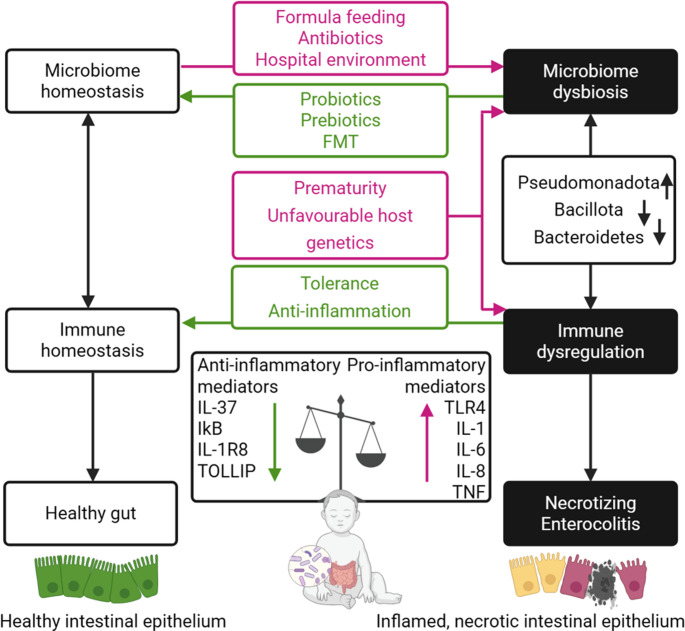 Figure 1
Figure 1- —Monash University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInfant Nutrition and Health · Gut microbiota and health · Preterm Birth and Chorioamnionitis
Introduction
Globally, necrotizing enterocolitis (NEC) affects 3–7% of very low birth weight (≤ 1500 g) infants younger than 32–33 weeks gestational age (GA) [1–3], increasing to 12% in infants weighing 501–750 g at birth [4]. First described in 1823 [5, 6], NEC was thought to originate from nosocomial infections until the mid-20th century. The term “necrotizing enterocolitis” was introduced in 1965 to describe ‘an often fatal syndrome in preterm infants consisting of vomiting, abdominal distention, gastrointestinal bleeding and shock’ [7]. Bell’s 1978 three stage NEC classification [8], which was updated in 1986 [9], still remains in clinical use today [10].
Due to its non-specific early presentation and unpredictable progression, NEC is difficult to diagnose and manage [11]. By the time of NEC diagnosis, infants often present with sepsis or multi-organ failure [12]. Treatment is limited to bowel rest, broad-spectrum antibiotics and supportive care (e.g. blood pressure management, intravenous nutrition). Mortality ranges from 20 to 30%, rising to 70% for surgical NEC [1, 13, 14]. Survivors face significant risks of neurodevelopmental delays and short gut syndrome [14].
Despite intensive research, NEC pathogenesis remains poorly understood, hindering prevention and development of novel treatments. Usually, infants are sterile in utero and acquire their microbiome from birth [15] in parallel with their immune system development. Although incompletely mapped, disrupted interactions between the immune system and the microbiome development are implicated in NEC. This review outlines the current knowledge of NEC-related changes in the immune system and microbiome development, and evaluates contributions made by pre-clinical in vivo and in vitro models in advancing knowledge of NEC.
Risk factors and general pathogenesis
Although the exact pathogenesis underlying the overwhelming inflammation and subsequent tissue injury that leads to NEC remains unclear, several risk factors have been identified: prematurity [16, 17], hypoxia [18], genetic predisposition [19], formula feeding [20], immature mucosal barrier integrity [21] and intestinal microbial imbalance (termed dysbiosis) [14, 22, 23].
Prematurity strongly associates with NEC incidence and severity [16, 17]: immature intestinal barrier function and motility potentially increase translocation of bacteria or their components, leading to excessive inflammation while the premature immune system is ill-equipped to combat pathogens in the extrauterine environment [23]. Single nucleotide polymorphisms in inflammatory cytokine receptors, including VEGFC−2578 A, IL-18−607 A and IL-4RA−1903 G, may increase inflammatory signaling and predispose infants to NEC [24]. Intermittent hypoxia from apnoea of prematurity (inversely correlated with GA) disrupts blood flow to the gut and increases the risk of tissue damage [18]. Formula feeding triples NEC risk [25], potentially due to the absence of protective compounds found in breastmilk (e.g. unbound free fatty acids [26], anti-inflammatory cytokines, immunoglobulins (Ig), growth factors, cellular components and commensal bacteria) [27, 28]. Moreover, the imbalance of the gut microbiome in NEC impairs immune homeostasis [29, 30] and will be one of the foci of this review.
NEC management
Attempts to improve NEC management, through changes in rates and timing of enteral feeding [31], administration of colostrum [32, 33], erythropoietin [34, 35], corticosteroids [36], arginine [37], Ig [38, 39] or lactoferrin [40] have shown little if any improvement in clinical outcomes. Probiotics have shown promise in reducing NEC, but have yet to reach their full potential, as discussed below.
NEC pathoimmunology
The pathoimmunology of NEC comprises innate and adaptive immune dysregulation [23]. As per the traditional paradigm, innate immunity dominates in early life, as the adaptive immune system slowly adjusts to the extrauterine environment [41].
Innate responses
In preterm infants, innate immune responses often lack counter-regulation, leading to uncontrolled intestinal cytokine production, tissue damage and reduced repair [35]. Tissue damage impairs the intestinal barrier and allows bacterial translocation, further increasing recruitment of innate inflammatory cells and augmenting inflammatory responses [42].
Pattern recognition receptors (PRRs)
Innate immunity is activated via pattern recognition receptors (PRRs), which detect pathogen associated molecular patterns (PAMPs) and trigger pro-inflammatory signaling [43]. Toll-like receptors (TLRs), a PRR subtype expressed by immune and intestinal epithelial cells, are key to distinguishing beneficial commensal bacteria from pathogens and are implicated in NEC pathogenesis [43]. Dysregulation, especially increased TLR4 or TLR2 and insufficient counter-regulatory TLR9 signaling, promoted inflammation in murine NEC [22], while inhibition of TLR4 or its signaling reduced NEC [44–46]. TLR4 is activated by lipopolysaccharide (LPS), a cell wall component of gram-negative bacteria [47] including Pseudomonadota (formerly Proteobacteria) which are increased in and may promote NEC [48, 49], though definite evidence is lacking. LPS-induced TLR4 signaling activates NF-κB-mediated production of pro-inflammatory cytokines (e.g. IL-1, IL-6, IL-8, TNF (tumor necrosis factor), IFN-γ (interferon-γ)) and also reduces the number of protective Tregs (regulatory T-cells) [14, 22, 50]. Other TLR4-mediated responses, including necroptosis (inflammatory programmed cell death) [51] and autophagy (lysosomal degradation of intracellular components) [52], may contribute to NEC pathogenesis. Notably, in Tlr4-deficient mice, reduced expression of the necroptosis-associated kinases Ripk1 and Ripk3 inhibited necroptosis and NEC-associated inflammation [51]. Autophagy is prototypically not associated with excessive inflammation; however, TLR4 induced autophagy can contribute to NEC development via inhibited enterocyte migration within the murine ileum [52]. The expression of TLR4 and TLR2 is also greater in human fetal enterocytes than mature cells, predisposing premature infants to greater inflammatory responses [53].
Muramyl dipeptide, a ligand of the intracellular immune receptor NOD2, inhibited Tlr4 expression in murine NEC, thereby reducing intestinal injury [54]. Expression of Tlr9 and Tlr4 were reciprocally related in murine embryonic development and TLR9 activation limited TLR4 signaling in enterocytes, preventing inflammation and NEC development [55]. NEC infant stool sequencing identified microbial community patterns supporting TLR4 activation and TLR9 under-stimulation [56]. Deficiencies in TLR4 signaling adaptors MyD88 and TRIF protected against murine NEC [35], while absence of Tlr2,* Tlr5* or Tlr9 increased NEC susceptibility, suggesting complex PRR interactions [57]. Moreover, deficiency in IL-1R8 (aka SIGIRR), a negative TLR signaling regulator which is also reduced in fetal enterocytes vs. adult cells, is also associated with NEC [22].
In summary, PRRs contribute to NEC pathogenesis, highlighting the role of environmental factors (e.g. microbial composition) in excessive inflammation characteristic in NEC.
Pro- and anti-inflammatory mediators
Gene expression of innate and adaptive immune response mediators is developmentally regulated [58], including via epigenetic mechanisms [59], predisposing preterm infants to excessive inflammation and NEC [22, 53, 59]. For example, in comparison to mature enterocytes, fetal cells have increased expression of pro-inflammatory genes such as TRAF6, NFKB1 and MYD88 [53], and increased TNF abundance in NEC tissues contributes to intestinal injury through apoptosis and reactive oxygen species [60].
Immature enterocytes also have lower abundance of IκB, an important NF-κB inhibitor that reduces pro-inflammatory mediators including IL-8 [61]. Further, in a rat NEC model, NF-κB blockade improved survival and tissue injury [62]. Anti-inflammatory genes, including TNFAIP3 and TOLLIP, were also reduced in fetal enterocytes compared to mature cells– and expression was even lower in enterocytes of NEC infants [53].
Anti-inflammatory cytokines like IL-10 and IL-37 reduce NEC severity in mice [63] and the intestinal IL-37 receptor is more abundant in non-NEC controls compared to NEC infants [22].
Overall, these findings suggest that developmental down-regulation of inflammation is disrupted in NEC, leading to excessive innate immune activation, partially initiated by intestinal TLR activation, production of pro-inflammatory mediators and impaired counter-regulatory anti-inflammatory mechanisms.
Adaptive responses
The adaptive immune system responds to specific antigens processed and presented by antigen-presenting cells, like macrophages. Upon antigen recognition and co-stimulation, naïve CD4⁺ T cells differentiate based on the antigen and local cytokine milieu. In NEC, reduced TGF-β_2_ led to increased intestinal macrophage infiltration and activation [64], potentially contributing to elevated T cell responses. Pregnancy skews the maternal and fetal/neonatal adaptive immune system towards type 2 polarization, protecting the hemi-allograft fetus from rejection [41]. In NEC, however, polarization shifts towards type 3, with increased IL-17 producing Th17 and type 3 innate lymphoid cells (ILC3s) [50] while IL-22 levels were reduced [22, 65]. Other sources of IL-22, such as unconventional CD8^+^ γδ T-cells, were also reduced in NEC [66]. Pro-inflammatory IL36A, IL36B, and IL36G were increased in NEC [22] and IL-36γ-deficient mice were protected from T-cell driven intestinal inflammation [67]. IL-36γ may also inhibit Treg development [68], and anti-inflammatory FoxP3^+^ Tregs were 60% lower in NEC infants’ ileal lamina propria compared to age-matched healthy controls [69]. Altogether, NEC is characterized by a skew towards inflammatory type 3 responses and a deficiency of anti-inflammatory Tregs, driven by dysregulated antigen processing, presentation, and T cell activation.
The intestinal microbiome in early life
During delivery and in the days and weeks after birth, the infant faces one of its biggest challenges: the first exposure to microbes. Despite reports of bacteria present in amniotic fluid [70], placenta [71], umbilical cord blood, fetal membranes [72] and meconium [70, 73], a comprehensive study of term and preterm placentas found that, except for Streptococcus agalactia, a pathogen that was detected in 5% of samples due to perinatal infection, bacteria were either acquired through contamination during labour/delivery or through laboratory reagents [74]. After birth, the intestinal microbiome diversifies and stabilizes during the first years of life, with commensal microbiota forming a symbiotic relationship with the host [75, 76]. Thus, to maintain health, a delicate balance between immune tolerance of commensal bacteria and effective control of pathogens must be achieved.
The intestinal microbiome in healthy infants
The initial colonizers of the healthy neonatal intestine include mainly facultative anaerobes [77, 78]. Between 3 and 12 weeks of life, the microbiome begins a period of diversification [79], including colonization with obligate anaerobes such as Clostridium,* Bifidobacterium* and Bacteroides species [75]. Gradual diversification of the microbiome continues until stabilization at around three years of age, when it resembles the adult microbiome [80].
In the first month after birth, the mode of delivery and the external environment (e.g. feeding, medications, locations) are the main drivers of microbiome composition [81]. Infants delivered through cesarean sections (C/S) are typically colonized by Enterococcus faecalis,* Enterococcus faecium*,* Staphylococcus epidermidis*,* Streptococcus parasanguinis*,* Klebsiella oxytoca*,* Klebsiella pneumoniae*,* Enterobacter cloacae* or Clostridium perfringens [76, 81]. For vaginally delivered infants, the intestinal microbiome is enriched in Bifidobacterium,* Escherichia* and Bacteroides [76, 81]. Prior to weaning, either full or partial breastmilk-feeding has the most pronounced effect on microbiome composition, with babies receiving breastmilk having a higher abundance of Bifidobacterium compared to exclusively formula-fed babies [76]. Maternal prenatal antibiotic therapy also impacts microbiome composition, with reduced Bacteroides vulgatus and increased Escherichia coli,* Klebsiella oxytoca*,* Klebsiella pneumoniae* and Enterobacter cloacae [81]. This is consistent with postnatal antibiotic administration, including reduced species richness after meropenem, cefotaxime and ticarcillin-clavulanate, and a dominance of multidrug-resistant members of Escherichia,* Klebsiella* and Enterobacter genera in infants [82]. Early alterations in diversity or relative abundance of microbes were associated with a broad range of later adverse health outcomes including obesity, inflammatory bowel disease, atopic disease, type 2 diabetes and behavioural disorders [83–85].
The intestinal microbiome in preterm infants
Since feeding establishment is often delayed in preterm infants [86], it should be considered as a common confounder when comparing the early-life microbiome to term infants. In comparison to term infants, preterm infants are reported to have an increased proportion of facultative anaerobes such as Enterobacteriaceae,* Enterococcaceae* and Lactobacilli, but fewer obligate anaerobes such as Bifidobacteria,* Bacteroides* and Atopobium [87]. At the class level, the bacterial composition in stool samples of very low birth weight preterm infants progressed from Bacilli to Gammaproteobacteria to Clostridia with age regardless of antibiotics, feeds or delivery mode, indicating that host biology also drives microbial colonization in the newborn [79]. Additionally, preterm infants showed increased abundance of potentially pathogenic hospital-associated bacteria including Escherichia coli,* Enterococcus faecalis* and Klebsiella pneumoniae, independent of birth weight, feeding mode or antibiotic treatment [88]. These findings support evidence that prolonged hospital stays, often unavoidable in most infants born < 35 weeks GA, substantially affect microbiome acquisition [81].
The intestinal microbiome in infants with NEC
It was first proposed in 1979 that the hospital environment may hinder normal microbial colonization and lead to NEC [89]. In 2001, this hypothesis was refined in recognition of data showing that colonization patterns may contribute to NEC [90]. Despite extensive research over the last decade as detailed in Table 1, no single microbe has been identified as the cause of NEC.
Applying culturing, 16S rRNA profiling and metagenomic sequencing of stool samples, most but not all [88, 91–95] studies identified differences in the intestinal microbiome across taxonomic levels between preterm infants with and without NEC. Increased Pseudomonadota [96–100] and Gammaproteobacteria [97, 101–103] alongside decreased Bacillota [97, 103, 104] and Actinomycetota [98, 100, 103, 105] are commonly reported in NEC. Many species have been reported as more abundant in or associated with NEC, including Clostridium spp. (C. perfringens,* C. butyricum*,* C. neonatale*) [102, 106–110], Klebsiella pneumoniae [106, 111–114], *Cronobacter sakazakii *(previously Enterobacter sakazakii, formula contaminate) [107], Citrobacter koseri [111], Corynebacterium striatum [115], Morganella morganii [115], Staphylococcus aureus [98, 102] and Enterococcus faecalis [114], while other genera are less abundant in NEC infants, including Lactobacillus [91, 99, 100, 111, 116, 117], Bifidobacterium [98, 100, 105, 113], Streptococcus [91, 99, 105, 114, 118] and Veillonella [118, 119]. Contradictory findings are common regarding the NEC microbiome. For example, while specific species such as K. pneumoniae, E. faecalis and Staphylococcus aureus associate with NEC, their genera Klebsiella, Enterococcus and Staphylococcus, respectively, are often less abundant in NEC infants compared to controls [102, 116, 120, 121].
These discrepancies could result from small sample sizes [115, 122, 123] or varying GA, bodyweight at time of sampling, NEC severity, feeding practices and use of medication between studies [93, 99, 103, 105, 114]. Sample collection protocols and procedural differences of microbiome analysis, including sample collection timing, 16S ribosomal RNA amplicon region and culture techniques, may also contribute to observation variations (Table 1) [124]. In addition to intrinsic bacteria factors, the timing of acquisition likely plays a role in determining the immune system’s reaction to specific microbes. Longitudinal studies have identified that the microbiome is less stable [125] and reduces in diversity prior to NEC development [104, 105, 111, 113, 123]. Thus, it is likely no single species is the driver of NEC, rather impaired patterns of microbiome acquisition and resulting dysbiosis alter inflammatory responses.
Table 1. Published studies on the intestinal microbiome in infants with NEC from 2015 to 2025 where n number is greater than one. Stool was sampled in all studies, except Romano-Keeler (2018) and Stewart (2019) in which frozen or formalin-fixed paraffin-embedded intestinal tissues were used, respectively. Abbreviations: necrotizing enterocolitis (NEC), ribosomal ribonucleic acid (rRNA), operational taxonomic units (OTUs), quantitative polymerase chain reaction (qPCR), denaturing gradient gel electrophoresis (DGGE), Temporal temperature gradient gel electrophoresis (TTGE), small intestine perforation (SIP)ReferenceClinical and procedural featuresObservations (compared to controls unless stated otherwise)GA at birth (weeks)Birth weight (g)Sample timen numbersTechnology usedSim(2015) [106]< 32< 1,250Pre diagnosis12 NEC44 controls 16S rRNA gene sequencing and cultureOverabundance of a Clostridium OTU and a Klebsiella OTU in NEC infants.Presence of Clostridium perfringens type A confirmed by culture in all samples in which Clostridium OTU was found.Zhou (2015) [107]24 to 31< 1,860Pre and post diagnosis12 NEC26 controls16S rRNA gene sequencingAt the genus level, Clostridium sensu stricto significantly higher in early onset NEC cases and Escherichia/Shigella and Cronobacter higher in late onset NEC cases.McMurty (2015) [118]≤ 34≤ 1,500Post or pre diagnosis21 NEC74 controls 16S rRNA gene sequencingTwo classes, Actinomycetota and Clostridia, significantly lower in NEC specimens.Abundances of two bacterial genera, Veillonella and Streptococcus, significantly lower NEC specimens across all three Bell’s stages.Cassir(2015) [108]28 to 321000 to 1750Post diagnosis15 NEC15 controls 16S rRNA pyrosequencing; culture-based methods and qPCRClostridium butyricum associated with NEC using molecular and culture-based methods or qPCR. Staphylococci less prevalent in NEC samples.Leach(2015) [115]24 to 32< 1,500Pre diagnosis4 NEC18 controls 16S rDNA gene sequencingNo significant difference in fecal communities between NEC and non-NEC infants.Potentially pathogenic bacteria, such as Corynebacterium striatum and Morganella morganii, detected significantly more in infants with NEC.Heida (2016) [109]24 to 29560 to 1735Pre diagnosis11 NEC22 controls 16S rRNA gene sequencingHigher abundance of Clostridium perfringens and Bacteroides dorei in meconium and stool samples a week prior to NEC diagnosis.Equivalent control samples were more dominated by Staphylococci.Abdulkadir (2016) [95]23 to 30500 to 1470Pre and post diagnosis10 NEC10 controlsqPCRNo difference in the bacterial load of stool samples collected from infants over a period of 2 weeks prior to and post NEC diagnosis.No difference in the bacterial load of stool samples from NEC infants and non-NEC infants.Warner(2016) [101]24.7 to 28.7≤ 1,500Pre diagnosis28 NEC94 controls16S rRNA gene sequencingAt the class level, increased proportions of Gammaproteobacteria and lower proportions of both Negativicutes and combined Clostridia–Negativicutes in infants who developed NEC.Ward (2016) [119]23 to 29415 to 1741Pre diagnosis27 NEC117 controlsShotgun metagenomic sequencingMicrobial diversity of NEC stool samples was similar to preterm controls prior to day 17 of life. After day 17 of life, NEC stool samples showed less Veillonella and increased Escherichia coli.Presence of E.coli in at least one stool sample correlated with NEC-associated death.Stewart (2016) [125]23 to 30500 to 1810Pre and post diagnosis7 NEC28 controls 16S rRNA gene sequencingThe microbial composition of stool from NEC infants is less stable than that of control infants.Barron(2017) [93]-≤ 1,500Pre diagnosis30 NEC65 controls 16S rRNA pyrosequencingNo significant differences evident between control and NEC, medically treated NEC, and surgically treated NEC and in pre-NEC and post-NEC samples.Roze(2017) [102]24 to 31-Post diagnosis16 NEC78 controls 16 S rRNA gene sequencingClostridium sensu stricto and Gammaproteobacteria associated with NEC.CultureAssociation between Clostridium at the genus level and Clostridium neonatale and Staphylococcus aureus at the species level with NEC.Lower proportion of Klebsiella and Citrobacter observed in NEC infants.Dobbler(2017) [111]≤ 32< 2,050Pre diagnosis11 NEC29 controls 16S rRNA gene sequencing, shotgun metagenomic sequencingCitrobacter koseri and/or Klebsiella pneumoniae are dominating taxa in NEC.Low diversity and low abundance of Lactobacillus correlate with NEC risk in preterm infants.Wandro (2018) [122]23 to 31620 to 1570Pre and post diagnosis3 NEC21 controlsqPCROverall microbial abundance was lower in NEC infant stool samples than controls. 16S rRNA gene sequencingNo microbial composition signature was linked to NEC.Itani(2018) [116]27 to 35< 2,250Pre and post diagnosis11 NEC11 controlsqPCR, TTGE and cultureNo significant difference in microbiota between NEC and non-NEC by culture method and TTGE.By qPCR, significant differences with a higher colonization by Staphylococci and lower colonization by Enterococci and Lactobacilli in NEC compared to healthy controls prior to NEC onset.Romano-Keeler (2018) [110]29 mean1,274Post diagnosis12 NEC14 controls 16S rRNA gene sequencingMicrobial diversity was lower in NEC tissue samples.Higher abundances of Staphylococcus and Clostridium sensu stricto, and lower abundances of Actinomyces and Corynebacterius in NEC.Stewart (2019) [96]25 median731 medianPost diagnosis13 NEC16 controls (SIP) 16S rRNA gene sequencingMicrobial diversity was lower in NEC tissue samples than SIP tissue samples.Pseudomonadota relative abundance was higher in NEC tissue samples.Bacteroidota relative abundance was higher in SIP tissue samples.Liu (2019) [123]28 to 33773 to 2149Pre and post diagnosis4 NEC17 controls 16S rRNA gene sequencingMicrobial diversity reduced from birth to NEC diagnosis.Control stool samples exhibited more stable microbiome composition than NEC stool samples pre-diagnosis.Enterococcus,* Streptococcus* and Peptoclostridium genera were more abundant in NEC stool samples.Feng(2019) [91]> 28< 2,700Post diagnosis16 NEC16 controls16S rRNA gene sequencingNo significant difference in the total diversity of microbiota between NEC cases and controls.Propionibacterium more abundant in NEC infants; Lactobacillus, Phascolarctobacterium and Streptococcus salivarius were more abundant in controls.Olm(2019) [112]28 to 32-Pre diagnosis34 NEC126 controlsGenome-resolved metagenomic analysisSignificantly more Klebsiella pneumoniae in NEC.Replication rates of all bacteria, especially Enterobacteriaceae family, significantly higher 2 days before NEC onset.Lindberg(2020) [97]23–27485 to 1,026Pre diagnosis5 NEC5 controls16S rRNA gene sequencingPseudomonadota and bacteria belonging to the class Gammaproteobacteria were more abundant among NEC infants. Control infants showed greater abundance Bacillota.Duan (2020) [113]≤ 37-Pre and post diagnosis28 NEC30 controlsPCR-DGGENEC stool samples had lower diversity.Bacteroides and Klebsiella was found in more NEC samples, whereas E.coli,* Bifidobacterium* and Lactobacillus were found in more control samples.Fu (2021) [117]≤ 33-Pre and post diagnosis15 NEC15 controls16S rRNA gene sequencingBacteroidota and Actinomycetota were more abundant at birth in infants that later developed NEC.Gammaproteobacteria, Enterobacteriaceae and Clostridiaceae are less abundant in stool of infants with diagnosed NEC.Shaw (2021) [56]< 32-Pre diagnosis11 NEC11 controlsqPCRNo difference in overall bacterial abundance between NEC and control samples.Shotgun metagenomic sequencingStool samples from NEC infants show either high proportions of LPS-expressing bacteria or low frequency of CpG motifs within their bacterial DNA.Tarracchini (2021) [114]23 to 39900 to 3010Post diagnosis64 NEC81 controlsShotgun metagenomic sequencingE.coli and Enterococcus faecalis were more abundant in stool samples from infants with NEC.Streptococcus agalactiae was more abundant in stool samples from control infants than those with NEC.Staphylococcus epidermidis and Klebsiella pneumoniae did not differ between NEC and control stool samples.Pre diagnosis26 NEC81 controlsKnown pathogen species K. pneumoniae,* Proteus mirabilis*,* C. perfringens*,* C. neonatale*,* Pantoea dispersa and S.aureus* were more abundant in stool samples prior to NEC diagnosis.Masi (2021) [98]< 32-Pre diagnosis14 NEC34 controlsShotgun metagenomic sequencingNEC stool samples showed higher abundance of Pseudomonadota and lower abundance of Actinomycetota.Enterobacter cloacae were more abundant, while Bifidobacterium longum was less abundant in NEC stool samples.Huang(2022) [99]--Pre and post diagnosis9 NEC10 controls 16S rRNA gene sequencingComposition differs between NEC and controls.In NEC infants the Pseudomonadota phyla was most abundant and there was reduced abundance at the genus level of Enteroccocus,* Streptococcaceae* and Lactobacillus.Du(2023) [100]≤ 34-Pre and post diagnosis16 NEC16 controls 16S rRNA gene sequencingWhile no difference in overall diversity was reported, at the phylum level abundances of Pseudomonadota increased and Actinomycetota decreased in NEC. At the genus level Bifidobacterium and Lactobacillus abundance decreased in NEC.Zenner(2023) [120]< 32922 meanPre and post diagnosis11 NEC21 controls 16S rRNA gene sequencingPrior to NEC diagnosis no difference was observed between NEC and controls. At day 21 of life, increased abundance of Escheria-Shigella and lower Enterococcus abundance seen in NECAires(2023) [94]< 32-Post diagnosis37 NEC66 controls 16S rRNA gene sequencingNo difference in abundance or diversity between NEC and controls.CultureAntibiotic susceptibility of C. butyricium,* C. neonatale*,* E. coli* strains isolated from NEC and controls did not differ.Wang (2024) [121]26 to 43700 to 4300Post diagnosis18 surgical NEC18 medical NEC18 controls 16S rDNA gene sequencingEnterobacteriaceae was enriched in intra-luminal stool collected from jejunum during NEC resection surgeries.Staphylococcus and Stenotophomas was enriched in intra-luminal stool collected from jejunum during control surgeries.Granulicatella was enriched in nappy stool collected from infants with NEC under medical management.Thanert (2024) [88]≤ 37≤ 1500Pre diagnosis48 NEC96 controlsShotgun metagenomic sequencing and transcriptional analysisNo metagenomic or transcriptional difference between NEC and controls microbiome. Persistent low diversity and functional immaturity is seen in only late-onset NEC (≥ 40 days of life) compared to controls.Chae(2024) [105]22 to 40-Pre diagnosis46 NEC/feed intolerance264 controlsqPCRSignificantly lower diversity in NEC/feeding intolerance infants despite no change in overall number of identified species. Lower abundance of Actinomycetota phyla, Streptococcus and Bifidobacterium genus in NEC/feeding intolerance infants.El Manouni El Hassani (2025) [104]≤ 30-Pre diagnosis56 NEC56 controlsIS-pro™C. perfringens was detected more often in NEC stool samples.There was no difference in abundance or diversity between stage II or IIIA NEC and control stool samples.Stage IIIB NEC stool samples were less diverse, with reduced Bacteroidota, Bacillota, Actinomycetota, Fusobacteria and Verrucomicrobiota.Wang (2025) [103]< 37-Pre diagnosis13 NEC13 controls 16S rRNA gene sequencingBacillota decreased and Gammaproteobacteria increased in the stool of preterm infants with NEC.
Intestinal dysbiosis and inflammatory responses in NEC
Intestinal dysbiosis has been suggested to precipitate inflammation and subsequently drive inflammatory diseases including NEC. This theory rests on findings that the intestinal microbiota differs between NEC and control infants, and that antibiotic treatment [126, 127], histamine 2 blockers [128], antacid therapy [129] and formula feeding [20], each known to alter the intestinal microbiome, increased the risk of NEC. Significant associative evidence suggests that one of the main drivers of excessive inflammation-induced tissue damage preceding NEC is the interactions between an abnormal microbiome and the preterm immune system (Fig. 1) [77]. Furthermore, a balanced stable microbiome has been linked to health and reduced inflammation, suggesting that microbiome instability can contribute to inflammation. While opportunistic pathogens such as Enterobacter cloacae [98, 130], E. coli [114, 119], Staphylococcus aureus [98, 102] and Klebsiella spp. [106, 111–114] have been associated with NEC, there is a lack of experimental validation demonstrating causation or functional mechanisms of microbiota-driven inflammation in NEC or model systems. The exceptions here are *Cronobacter sakazakii *and Clostridium species, which are discussed in further detail below.
Fig. 1. Microbiota-mediated immunopathogenesis of NEC. Risk factors in early life can lead to gut dysbiosis, increasing the abundance of TLR4, pro-inflammatory cytokines and inflammatory mediators, resulting in inflammation. Due to a prematurity-associated reduced abundance of IκB, IL-1R8 and TOLLIP, inflammation can quickly get out of control leading to the inflammation and tissue damage seen in NEC. This is not a comprehensive list of inflammatory mediators and work is ongoing to characterize the pathoimmunology of NEC. Red arrows/boxes indicate harmful effects and green arrows/boxes indicate beneficial effects in NEC. Created in BioRender. Abbreviations: necrotizing enterocolitis (NEC), fecal microbiota transplant (FMT).
Inflammation induced by Cronobacter sakazakii
In 1998, a NEC outbreak arose from Pseudomonadota species, C. sakazakii, via formula contamination [131]. Experimental validation of C. sakazakii infection in murine NEC revealed that increased dendritic cell (DC) activation and decreased macrophage/neutrophil recruitment in the intestine precipitated NEC [132, 133]. In vitro infection with C. sakazakii led to increased abundance of IL-1β, TLR4, the inflammasome components NLRP3 and caspase-1, and the adapter protein MyD88 in human colon HT-29 cells [134]. C. sakazakii also induced cell death in jejunal J774A.1 enterocytes and intestinal damage in murine NEC [134]. Neutrophil- and macrophage-mediated phagocytosis conferred protection against C. sakazakii, their depletion from the intestine lamina propria worsened murine NEC and was accompanied by increased abundance of DCs, pro-inflammatory cytokines and inducible nitric oxide synthase [133].
Inflammation associated with Clostridium species
Clostridium species of the Bacillota phylum, such as C. neonatale,* C. butyricum* and C. perfringens, may contribute to NEC pathogenesis [108, 128, 135, 136], although the mechanistic pathways through which Clostridium species promote intestinal inflammation remain elusive. C. perfringens abundance was increased in NEC piglets [137] and activation of TLR4 by Clostridia may occur in NEC as infection induced host immune responses involving TLR4/MyD88/NF-κB signaling pathways in piglets [138]. Complementing in vitro experiments showed induction of the pro-inflammatory CCL5 and IL8 and the anti-inflammatory IL1RN, NFKBIA and TNFAIP3 due to activation of TLR2 by C. perfringens’ cell wall component peptidoglycan when incubated with intestinal porcine IPEC-J2 enterocytes [137]. Heat-killed C. butyricium and C. neonatale isolated from NEC infants induced IL-8 in human Caco-2 colon cancer cells, but the same bacteria isolated from control infants did not [139].
Insufficiency of anti-inflammatory responses
Actinomycetota, Bacillota and Bacteroidota are reportedly less abundant in NEC compared to non-NEC infants [30, 97, 112, 140–143]. Despite an overall reduction in Bacillota abundance, the enrichment of certain Clostridium species may promote NEC pathogenesis, indicating the complexity of the role of microbiota in NEC. Phylum level classification is too coarse to discern specific inflammatory properties. Bacteria from the Bacillota, Bacteroidota and Actinomycetota phyla can initiate inflammatory pathways through their microbial-associated molecular patterns, but also induce Tregs and other regulatory mechanisms, possibly dampening inflammation and augmenting tolerance [144, 145]. Low level production of short chain fatty acids (SCFAs) by bacteria, including Bifidobacterium infantis, also contributes to mucosal integrity [146].
Modeling NEC
Studying NEC in human infants is not straightforward. Only minimal blood volumes and small intestinal resection specimens can be obtained. Stool usually collected from nappies, though more accessible, may not accurately represent the microbiome at the site of disease. Thus, modeling NEC is key to advancing knowledge.
Most current NEC pathogenesis evidence stems from animal studies in rodents, rabbits, pigs, and non-human primates (NHPs) [23, 147]. Although animal models provide important mechanistic insights, it is critical to understand their limitations in terms of human disease transferability, including differences in intestinal development, enteral nutrition, disease pathology [147] and particularly microbial composition [148, 149]. Caution is needed in interpretation of animal microbiome data, as the overlap with the human microbiome varies (e.g. only 3–4% overlap between humans and mice) [148, 149]. Furthermore, replicating the multifactorial origins of NEC in animal models is challenging.
In vivo models
Rodents
In 1974, Barlow and colleagues used newborn rats as the first animal model of NEC. Control pups received rat breastmilk for 72 h, while NEC was induced via formula feeding combined with daily 3–5 min asphyxia, with or without Klebsiella pneumoniae gavage-inoculation [150]. Asphyxia mimicked hypoxic episodes, common in preterm infants. The rat model reproduced human NEC symptoms, including abdominal distention, lethargy, bloody stools, and necrotic intestines [150] but lacked hallmark features of human NEC, pneumatosis intestinalis or portal venous gas. To mimic vasoconstriction and ischemic-reperfusion injury known to NEC, modifications such as 5 min of 100% O_2_ reoxygenation followed by asphyxia [151] and 5 min of cold stress at 1°C were included [152].
Subsequent NEC rat models applied hypoxia (usually 5% O_2_/95% N_2_ for 3–10 min) [153–156] in combination with formula feeding to induce NEC-like lesions in the intestines of preterm, term and 2-3-day old rats [153–156]. Since NEC was often mild in this model, LPS administration was added to mimic intrauterine inflammation via TLR4 activation [157]. This three-hit-model enabled testing of microbiome-modulation, e.g. human milk oligosaccharides (HMOs) [158], fecal microbiota transfer (FMT) [159] or administration of probiotic Lactobacillus reuteri, Bifidobacterium longum infantis [160], Bifidobacterium adolescentis [161] and Bifidobacterium bifidum [147], with varying clinical outcomes.
Despite the challenge of smaller pups, mouse NEC models have gained popularity over rat models due to the advantage of using genetically modified strains. Notably, term murine pup intestines are immature at birth, and only reach full maturity by postnatal day 28 [162]. NEC-like pathology has been introduced in newborn [22, 148], 3-day [163], 7-10-day [164, 165] or in 20-36-day old [166] mice via exposure to hypoxia/asphyxia, cold stress and formula feeding. NEC pathogenesis increased with feeding formula spiked with LPS or stool-derived bacterial slurry from NEC-infants, reinforcing the role of microbial dysbiosis.
In summary, rodent models of NEC have improved our understanding of the microbiome’s role in NEC pathogenesis and potential treatments. However, inconsistencies between models and inter-species microbial differences limit the application of those findings.
Other small animals
While rodents represent the most employed small animal NEC model, rabbits, hamsters, chickens and gnotobiotic quails are also available [167]. Rabbits require the small intestine or colon to be surgically segmented and injected with casein and calcium gluconate to mimic human NEC [168–170]. This time- and labour-intensive model was used to link NEC with low pH [169], low lactase, organic acids, and high levels of histamine and O_2_-derived free radicals [171, 172]. Other experiments involving preterm C/S-delivered rabbits have shown that formula supplemented with Enterobacter cloacae combined with poor intestinal motility induced NEC-like pathology [173].
Less common are germ-free quails and chickens to study specific bacterial species isolated from human NEC stool [174, 175]. Oral inoculation of Clostridium beijerinckii, in 2-50-day old axenic chickens [174] caused cecal inflammation and necrosis. Gnotobiotic quails orally inoculated with Clostridium butyricium, which produces butyric acid, developed intestinal NEC-like lesions [175].
Large animals
Piglets delivered at ~ 90% full gestation (104 days) represent an attractive model for NEC due to gastrointestinal maturity similar to preterm infants born at 30–32 weeks GA [176]. In both term and preterm piglets, intestinal intraluminal casein injection induced NEC-like lesions [177, 178]. In term piglets, mesenteric artery and lymphatic occlusion caused necrosis, immune infiltration, mucosal thinning and epithelial barrier disruption, mimicking human NEC [179], as did a combination of hypoxia and cold stress [180]. In weanling pigs, intraperitoneal LPS injection or formula supplementation induced intestinal epithelial necrosis reminiscent of NEC [181]. These studies implicated diet and microbial fermentation of undigested carbohydrates (producing CO_2_ and H_2_), as causes of pneumatosis intestinalis and portal venous gas in NEC [182].
Non-human primates (NHPs) most closely resemble humans in modelling NEC. However, NHP models are extremely expensive and rarely employed in NEC research [147]. Baboons, delivered preterm by C/S, maintained in intensive care using human protocols and fed infant formula for 21 days, developed NEC at an incidence of 5% [64], with a similar clinical presentation as babies affected by NEC (abdominal distention, intestinal inflammation, haemorrhage). As in human preterm infants, intestinal TGF-β_2_ was decreased in preterm vs. term baboons and further decreased in NEC [64]. Additionally, NHPs and piglets share microbiome similarities to humans at a phylum level following birth [183, 184].
In summary, NHPs followed by piglets come closest to human infants in terms of intestinal maturity, physiology, nutritional requirements and likely also their microbiome and immune system development and function. However, large animal models carry high costs and require specialized facilities and training, rendering their use unfeasible for many scientists.
In vitro and ex vivo models
Cell culture
Barrier function, inflammatory signaling and cell-cell or bacteria-cell interactions have been investigated in Caco-2 and HT-29 adult human colon carcinoma epithelial cells, H4 human fetal small intestinal cells, IEC-6 rat small intestinal epithelial cells and IPEC-J2 jejunal porcine enterocytes [137, 185]. Caco-2 monolayers were used to study translocation of two early intestinal colonizers, E. coli, which was increased in immature, undifferentiated cells and C. difficile mediated translocation through toxin B opening of tight junctions [186, 187]. Although valuable to investigate specific cellular responses and barrier functions, single cell-type in vitro cultures cannot mimic crosstalk between immune, epithelial, and endothelial cells in vivo and immortalized cell lines often have altered immune responses and barrier functions when compared to primary cells.
Primary tissue xenografts
Primary human fetal small intestinal tissue without mesentery can be subcutaneously implanted into young immunocompromised mice and harvested at different maturity stages for organ culture [53, 188, 189]. Xenograft models have significant benefits over single cell culture: they are generated from primary human tissue, have relevant immune responses and maintain structural integrity important for microbiota interaction [53]. Following LPS- or IL-1β-stimulation, exposure to Bifidobacterium infantis- and Lactobacillus acidophilus-conditioned media attenuated excessive IL-8 and IL-6 production [188], likely via reduced TLR4/MyD88-mediated NF-κB activation, indicating potential beneficial effects of probiotics [188].
Organoids, organoid-microbiota co-cultures, and organ-on-a-chip
Organoids are 3D-structures grown from stem cells that self-organize through cell sorting and spatially restricted lineage commitment [190]. Unlike primary tissue explants, they survive in long-term culture (weeks) and intestinal organoids spontaneously form an intact epithelial layer and a crypt-villus structure. Organoids from pluripotent stem cells, fetal intestines and infant tissue resections resemble immature intestines and can be used to model NEC [191].
Intestinal organoids from mouse pups exposed to hypoxia plus LPS or LPS alone for 48 h [192, 193] exhibited increased Il6,* Tnf* and Tlr4 expression and disrupted tight junctions [192, 193]. Breastmilk components, including HMOs, exhibited protective effects in these organoid NEC models [191, 193]. In human neonatal intestinal organoids, LPS increased TNF,* IL1B*,* TLR4* expression and apoptosis [194, 195], while microbial SCFAs reduced inflammation in IL-1β-stimulated fetal small intestinal organoids [196].
Further, organoids can be co-cultured with bacterial species from NEC infant stool, e.g. uropathogenic E. coli [197] and Akkermansia mucinophilia [198] species, by micro-injection into the organoid lumen, enabling interaction with epithelial cells [199]. This enables functional studies of specific bacteria or stool-isolated bacterial communities and their role in NEC pathology.
The novel microfluidics-based gut-on-a-chip (GoC) technology models the in vivo-intestinal micro-environment by creating separate luminal and submucosal compartments. GoC facilitates co-cultures with bacteria and immune cells via microfluid channels [200, 201] without needing to micro-inject bacteria. GoC models are promising to study inflammation and host microbiome interactions with commensal bacteria in NEC.
NEC model summary
Various NEC models, ranging from low-throughput large animal models to small animal models, high-throughput cell line studies and advanced in vitro models such as GoC offer unique advantages for studying different disease aspects. However, when investigating the microbiome in NEC, substantial cross-species differences must be considered. Microbiome data from non-human models that do not use gnotobiotic approaches or fail to look beyond microbial diversity must be interpreted cautiously.
Modulating inflammatory responses in NEC using microbiota-targeted intervention
Several beneficial microbes have been investigated to prevent or ameliorate NEC outcomes. Strategies, such as administering prebiotics, probiotics, FMT or live biotherapeutics, aim to achieve an anti-inflammatory and tolerogenic intestinal environment.
Prebiotics
Prebiotics are defined as “a substrate that is selectively utilized by host microorganisms conferring a health benefit” [202]. Notably, human milk has prebiotic properties; it is rich in bioactive molecules, such as carbohydrates, proteins, fats, cytokines and Ig, that promote healthy microbial colonization and favourably regulate immune and inflammatory responses [203]. The risk of NEC can also be reduced by feeding breastmilk to neonates [204]. Beneficial effects include inhibiting signaling via TLR2 (soluble TLR2, soluble CD14), TLR4 (soluble CD14, lactadherin, lactoferrin, 2’-fucosyllactose), TLR3 (3’-galactosyllactose) and TLR7 (β-defensin 2) [203]. Meta-analyses have found that while some prebiotics (oligosaccharides, inulin, lactulose, lactoferrin) reduced mortality and rates of sepsis, there was no significant difference in NEC morbidity [40, 205]. Other prebiotic components of human breastmilk (e.g. maternal Ig, HMOs, and arginine) have also been suggested to reduce NEC severity and to alter microbial composition [206–209]. While many other components of breast milk likely have prebiotic effects, here we will focus on the microbiome and immune modulation induced by HMOs and maternal Ig.
Human milk oligosaccharides (HMOs)
After lactose and lipids, HMOs are the third-most abundant ingredient in human breastmilk, 13% of which are sialylated-HMOs (sHMOs) [210]. In colostrum, HMOs are more concentrated (20–25 g/L) than in mature milk (5–15 g/L) [211], however, infant formula only contains small amounts (100 mg/L) [212]. Many HMOs are indigestible for infants [213], but as the predominantly protective Bifidobacteria and Bacteroides have enzymes to metabolize HMOs, they selectively support their own intestinal colonization [214]. Thus, the low HMO quantities in formula were proposed to contribute to microbial dysbiosis and inflammation in formula-fed infants.
Approximately 200 structurally different HMOs are known, however, the functional role of different HMOs are largely unknown. Their composition in breastmilk is individual to each mother and varies throughout the lactation period. The best studied HMO is 2′-fucosyllactose, as it is found in 80% of all women’s breastmilk at a concentration of ~ 2.5 g/L. Microbial communities of young children incubated with pooled HMOs showed significant compositional shifts, including increased abundance of potentially beneficial Bacteroides, providing evidence that HMOs can directly alter the microbiome [215]. Animal models of NEC concluded that sHMOs may help to reduce the risk of NEC in premature infants [216]. Administering HMO-containing formula decreased proinflammatory markers, including CXCL15 (murine IL-8 homolog), in the serum and ileum of NEC-affected mice [206], reduced TLR4 protein abundance, NF-κB signaling, and restored proliferation of intestinal crypt cells in the ileum and in organoids [206]. Further, the NEC incidence in rats was reduced by sHMOs alongside TLR4, NLRP3 and caspase-1 levels [158].
Importantly, sHMO concentrations are lower in NEC infants’ compared to healthy infants’ maternal milk [98], and increased numbers of NLRP3- and caspase-1-positive lamina propria cells were observed in intestinal biopsies of NEC patients [158]. These findings suggest that sHMOs support the development of a beneficial microbiome, reduce inflammation and prevent NEC.
Maternal immunoglobulin (Ig)
IgA antibodies, secreted into the intestinal lumen and into breastmilk (secretory (s)IgA) [217], influence the composition of the intestinal commensal microbiota and bind to and protect against pathogens by preventing their recognition by TLRs [144, 217]. The intestinal sIgA abundance is lower in preterm infants than in their term peers. However, there is ample sIgA in maternal milk [144, 218], and it was surmised that the lower abundance of sIgA in formula-fed preterm infants predisposes them to NEC [217, 219]. Indeed, the lack of sIgA in formula promotes the activation of inflammatory pathways in response to bacterial colonization in preterm infants, thus likely driving NEC [53, 59, 188]. Notably, human NEC was associated with a reduction in IgA-bound bacteria; and most non-IgA-bound bacteria belonged to the potentially harmful Enterobacteriaceae family [207]. The lack of IgA coverage of pathogenic bacteria allows increased interaction with the reactive preterm immune system and bacterial translocation, potentially driving more severe inflammation. As seen in a murine NEC model, wild-type pups fed by IgA-deficient dams were more susceptible to NEC than those fed by IgA-producing dams, with NEC pathology similar to that of formula-fed wild-type pups [207]. Notably, Gammaproteobacteria-specific IgA partially facilitated the transition from premature to mature intestinal microbiota in mice [220].
In addition to IgA, maternal IgG provides passive immunity against enteric pathogens while humoral immunity develops, but is transferred mainly during the last trimester of pregnancy [221]. Hence, preterm infants are deprived of the protective capacities of IgG [221]. However, in a clinical trial, oral IgG failed to reduce NEC incidence [38]. Likewise, in another clinical trial, formula supplemented with IgA/IgG (73%/26%) failed to reduce NEC incidence and severity in human neonates [222], and oral gentamicin was found to be more effective at preventing NEC compared to oral IgA/IgG administration alone [223]. One major limitation of these trials was, however, that the effect of sIgA [217], which, due to the secretory component is more stable in the luminal environment, was not tested. Likewise, no clinical trial to date has evaluated the effects of IgA alone.
Probiotics
Probiotics are defined as “live microorganisms which when administered in adequate amounts confer a health benefit on the host” [224], however, most probiotics do not colonize the intestine themselves but rather support the growth of other beneficial bacteria by improving barrier function and anti-inflammatory responses [225]. Studies in very (< 32 weeks GA) and extremely (< 28 weeks GA) preterm infants have shown that probiotic administration alters microbial diversity and composition [226, 227]. Probiotics vary in composition, often containing only a few microbial strains, and are rather simplistic compared to the vast diversity of species that colonize the infant’s intestine [225]. Lack of standardization and strain-level characterization also often creates batch-to-batch variation, complicating interstudy comparisons, including the interpretation of meta-analyses [228]. Despite some randomized controlled studies reporting minimal benefit of probiotics [229–232], most prospective cohort studies [233, 234], retrospective studies [235–237], randomized controlled trials [238–240] and meta-analyses [229, 241–244], predominantly investigating the effects of Lactobacilli, Bifidobacteria and Bacteroides, have suggested that probiotics do reduce the incidence of NEC. Further details regarding the anti-inflammatory effect of probiotics on the immune system in NEC models can be found in Table 2. Overall, current understanding is impeded by substantial heterogeneity regarding strains, dose, mode and timing of administration and outcomes evaluated. Considering these circumstances, it is unsurprising that evidence is somewhat conflicting.
In 2021, the American Academy of Pediatrics published a clinical report recommending against routine administration of probiotics in preterm infants, especially when birth weight is < 1000 g, citing concerns about the lack of a Food and Drug Association (FDA)-approved probiotic alongside conflicting safety and efficacy data [245]. Following a report of fatal Bifidobacterium longum probiotic sepsis, the FDA also released a statement warning of invasive disease risk in preterm infants receiving probiotics [246]. In response, the European Society of Paediatric Gastroenterology, Hepatology and Nutrition, alongside the European Foundation for the Care of Newborn Infants, acknowledged the need for future high-quality research, while advocating for the continued use of probiotics in preterm infants as documented benefits outweigh reported rare adverse effects [247]. A comprehensive 2025 meta-analysis of probiotic use including 20,323 preterm infants found probiotic sepsis occurred at a rate of less than 0.04% and NEC incidence decreased (relative risk 0.60, 95% confidence interval 0.51–0.71) [248]. Despite documented benefits, research into how probiotic species interact with the immune system to prevent inflammation is ongoing. Thus, the concept of probiotic treatment likely has not yet achieved its full potential.
Table 2. Pre-clinical evidence of probiotics modulating inflammatory responses from 2013 to 2024. Different probiotic strains were administered to mice, pig, rats, or cultured cells with or without inflammatory stimulation with produced changes in mRNA expression, protein abundance and/or cell proportions. Abbreviations: lipopolysaccharide (LPS), interleukin (IL), messenger ribonucleic acid (mRNA), single Ig IL-1-related receptor (SIGIRR), toll interacting protein (TOLLIP), regulatory T-cells (Tregs), toll-like receptors (TLR), nuclear factor kappa-light-chain-enhancer of activated B-cells (NFκB), chemokine (C-X-C motif) ligand 1 (CXCL), nitric oxide synthase (NOS), tumor necrosis factor (TNF), interferon (IFN), interleukin-1 receptor-associated kinases (IRAK), primary intestinal epithelial cells (P-IEC)Probiotic strainsHost speciesSample typeInflammatory stimuliModulation of immune mediators in response to probioticsMechanism of action of probioticsReferencesIncreasedDecreasedBifidobacterium infantis and Lactobacillus acidophilus-conditioned mediaHumanImmature human intestinal xenograftsLPS + IL-1αmRNA: SIGIRR and TOLLIPmRNA: TLR2, TLR4Protein: IL-8, IL-6-Ganguli (2013)[188]HumanPrimary enterocyte culture of NEC tissueLPS + IL-1αmRNA: SIGIRR and TOLLIPmRNA: TLR2, IL8, IL6TLR2-mediated anti-inflammatory activityDNA from Lactobacillus rhamnosus HN001MouseIleumFormula + hypoxia-mRNA: Nos2TLR9-mediated reduction of NEC severity and inflammationGood (2014)[165]PigletIleumFormula-mRNA: NOS2-HumanIleumLPS -mRNA: NOS2, IL6; NF-κB translocationTLR9-mediated inhibition of TLR4 signalingBifidobacterium longum subsp. infantisRatIleumFormula + hypoxia + cold stress-mRNA: Il6, Cxcl1, Tnf, Il23Protein: iNOS-Underwood (2014)[160]Lactobacillus reuteri DSM 17938MouseIleumFormula + hypoxia + cold stressTregsEffector/memory T cells-Liu (2014)[249]Bifidobacterium microcapsulesRatIleumLPS + formulaProtein: TLR2, TLR4, NF-κB p65-Zhou (2015)[250]Bifidobacterium longhum subsp infantis conditioned mediaHuman and mouseH4 cell line; human fetal small intestinal xenografts;mouse fetal ileum; primary NECenterocytesIL-1β-mRNA: IRAK2Protein: IL-6; phospho-c-Jun and phospho-c-Fos (in H4 cells)TLR4-mediated anti-inflammatory activityMeng (2016)[251] Bifidobacterium adolescentis Rat IntestineFormula + asphyxia + cold stressmRNA: Tollip,* SigirrmRNA: Tlr4-Wu (2017)[161]Bacteroides fragilis and/or PSAMouse IntestineIL-1β-Protein: IL-8-Jiang (2017)[252]PSA of B. fragilisHumanH4 cell line; fetal enterocytes from resected NEC smallintestineIL-1β-Protein: IL-8TLR2- and TLR4-dependent inhibition of AP-1 genes JUN and FOS Jiang (2017)[252]Lactobacillus reuteri DSM 17938MouseIleum; spleen (DCs)Formula- + hypoxia + cold stressFoxP3^+^ Tregs;Tolerogenic DCs% of activated effector CD4^+^ T cellsProtein: IL-8IFN-γ, IL-1βTLR2-mediated DC recognition and DC-priming of TregsHoang (2018)[253]PSA of Bacteroides fragilisHumanH4 (fetal small intestinal cell line)IL-1β-mRNA: IL8Protein: IL-8Zona pellucida protein 4 (TLR2- and TLR4-dependent)-mediated decrease in expression of IL8Gorreja (2019)[254]Lactobacillus reuteri DSM 17,938MouseSmall intestine--FOXP3^+^ Tregs-Liu (2019)[255]Bacteroides fragilis strain ZY-312RatSerum; faeces Cronobacter sakazakii Protein: EGF (serum);Protein: TNF, IFN-γ (serum);-Fan (2019)[256]ILA from Bifidobacterium longhum subsp infantisHuman and mouseH4 cell line; intestine (mouse)IL-1β-Protein: IL-8, MIP2Aryl hydrocarbon receptor mediated anti-inflammatory responseMeng (2020)[257] Lactobacillus rhamnosus GG MouseIntestineFormulamRNA: Sigirr, A20mRNA: Icam1, Il1b*,* Il8*,* Tlr4*,* Irak1Induced expression of TLR inhibitors inhibits TLR-dependent inflammationCuna (2021)[258]Bifidobacterium infants, Lactobacillus acidophilus*,* Enterococcus*, and Bacillus cereusMouseIleumFormula + hypoxia + cold stress-mRNA: Tnf,* Il1b*,* Il6PXR suppression of JNK signaling enhances barrier functionZhao (2021)[259]ILA from Bifidobacterium longhum subsp infantisHuman and mouseH4 cell line; intestine (mouse)IL-1βmRNA: Il6, Stat1/STAT1mRNA: Il8, Paf1*,* Pafr*,* Traf2Upregulation of Stat1/STAT1 inhibits IL-1β driven inflammationHuang (2021)[260] Ligilactobacillus salivarius YL20 MouseSmall intestine Cronobacter sakazakii -mRNA: Il1b, Il6*,* TnfProtein: IL-1β, IL-6, TNF-Wang (2022)[261]Lactobacillus rhamnosus or Lactobacillus acidophilus* and Bifidobacterium bifidumHumanCord blood mononuclear monocytesLPS% of TLR4 + monocytes, Protein: IL-1β, TNF-Stimulation of immune cells may improve their ability to fight infectionRückle (2022)[262]Lactobacillus rhamnosus GGMouseIleumPrenatal antibiotics + formulamRNA: Gpr81,* Axin2*,-Activation of Wnt signaling increases intestinal proliferationCuna (2023)[263] Bifidobacterium breve AHC3 RatIleumFormula + LPS + hypoxia + cold stressProtein: IL-10Protein: TNFInhibition of iNOS expression protects against intestinal injuryLin (2023)[264] Lactobacillus reuteri DSM 17938 MouseIleumFormula + hypoxia + cold stress -mRNA: Tnf,* Il1bProtein: TNF, IL-1βImproves gut barrier function, by reduced pro-inflammatory cytokine productionLai (2024)[265] Bacteroides fragilis RatColonLPS + hypoxia + cold stressmRNA: Il6Protein: FXR, NLRP, IL-1βBacteroides fragilis* metabolizes bile acids and inhibits FXR-NLRP pathwayChen (2024)[266] Limosilactobacillus fermentum CECT5716 HumanP-IECsLPS-Protein: IL-6TLR9 mediated anti-inflammatory activityHedegger(2024)[267]
Lactobacillus spp
L. reuteri increases tolerogenic DC and FoxP3^+^ Treg numbers, while reducing effector CD4^+^ T cells and IFN-γ and IL-1β abundance by inhibiting TLR2 [253], thus reducing murine NEC severity. Likewise, increased intestinal Foxp3^+^ Tregs following administration of L. reuteri strain DSM-17938 was suggested to confer protection against NEC by decreasing the mRNA and protein abundance of Il6,* Tnf*,* Tlr4* and Nfkb, while increasing anti-inflammatory Il10 in the intestine of NEC-affected rats (Table 2) [255]. Feeding L. reuteri to healthy dam-fed mice increased bacterial diversity and relative abundance of the Clostridiales order and decreased relative abundance of the genera Bacteroides,* Sutterella*,* Akkermansia* and Ruminococcus compared to controls [255]. However, how changes in relative bacterial abundance facilitate or prevent NEC remains unclear, pointing to the limited microbiome overlap between human and mice [149], especially as many Lactobacilli species only colonize the human intestine in the absence of competition or following microbiome perturbation. Nevertheless, a randomized, double-blinded, placebo-controlled trial showed that L. reuteri supplementation caused increased bacterial diversity and reduced abundance of Enterobacteriaceae and Staphylococcaceae in extremely low birth weight infants, however, no significant positive effect on NEC incidence was reported [268].
Similarly, the Lactobacillus rhamnosus strain HN001 reduced NEC severity in mice and piglets [165]. In intestinal tissue from humans and in cultured IEC-6 rat enterocytes, Lactobacillus rhamnosus HN001 inhibited TLR4 signaling via TLR9 activation (Table 2) [165]. Lactobacillus rhamnosus strain GG reduced the intestinal injury severity in the colon of NEC-affected mice by augmenting IL-10 receptor-mediated signaling, causing STAT3-mediated induction of SOSC3, which prevented pro-inflammatory TNF and CCXL2 protein production [269].
Bifidobacterium spp
B. adolescentis reduced NEC severity in rats by decreasing Tlr4 (Table 2) [161]. Similarly, Bifidobacterium infantis-conditioned medium or Bifidobacterium microcapsules decreased intestinal TLR2 and TLR4 mRNA in immature human intestinal xenografts [188], or their protein abundance in rats (Table 2) [250]. Interestingly, TLR4 was required for the inhibition of IL-6 by B. infantis-conditioned medium in enterocytes isolated from human NEC tissue and fetal murine intestinal tissue [251]. Being gram-positive, Bifidobacteria lack LPS but are thought to modulate TLR4 via yet unidentified secretory molecules [251]. In term newborn NEC rats, microcapsules of Bifidobacterium also reduced the abundance of NF-κB p65, while B. longum infantis reduced intestinal expression of the pro-inflammatory Il6,* Cxcl1*,* Tnf*,* Il23* and Inos (Table 2) [160]. In immature human intestinal xenografts and primary enterocyte cultures of NEC tissue, B. infantis- and L. acidophilus-conditioned media also increased anti-inflammatory mediators IL1R8 and TOLLIP, and reduced IL-6 and IL-8 protein concentrations in a TLR2- and TOLLIP-dependent fashion (Table 2) [188]. Similarly, B. adolescentis reduced NEC severity by increasing Il1r8 and Tollip mRNA in a rat NEC model [161]. A lack of anti-inflammatory mediators including IL-1R8 and TOLLIP leading to excessive inflammatory responses of NEC-affected epithelia is further supported by our own study in which intestinal IL-1R8 was decreased in NEC infants compared to healthy controls and infants that had recovered from NEC [22].
B. infantis found in breastmilk can also secrete the anti-inflammatory tryptophan metabolite ILA (indole-3 lactic acid) [257, 260] that reduced IFN-γ in immature human enterocytes and the colon and ileum of mouse pups [260]. Thus, ILA might also mediate the anti-inflammatory effects seen in response to B. infantis-conditioned media.
Bacteroides spp
The developmentally regulated gene ZP4 (zona pellucida-protein 4) was implicated in the anti-inflammatory functions of Bacteroides fragilis’ surface component PSA (polysaccharide A), reducing IL-8 concentrations in H4 fetal intestinal cells in a TLR2- and TLR4-dependent manner (Table 2) [254]. Moreover, pretreatment with the B. fragilis strain ZY-312PSA ameliorated NEC severity and intestinal injury caused by C. sakazakii infection by inhibiting pro-inflammatory TNF and IFN-γ and the inflammasome (NLRP3, caspases 1/3), while increasing the anti-inflammatory IL-10 [256]. In rats, B. fragilis ZY-312 pretreatment, furthermore, prevented C. sakazakii-induced harmful microbiome changes including reduction of Bacteroidota and increase of Pseudomonadota species associated with human NEC (Table 2) [256].
Fecal microbiota transfer (FMT) and fecal filtrate transfer (FFT)
Transfer of intestinal microbiota-containing fecal material from a healthy donor to a sick individual (rectally, orally or via a stoma) as whole fecal material (FMT) or filtered as FFT, leaving only bacteriophages, extracellular vesicles and bacterial products, has shown promise in numerous gastrointestinal conditions [270]. FMT is an established treatment for C. difficile infections [271], and has been suggested for the treatment of Ulcerative colitis [272], Crohn’s disease [271], insulin resistance [273] and NEC [159, 274–277]. In a rat NEC model, FMT from a healthy adult to a neonatal rat decreased IL-1β, TNF and IL-6 and intestinal neutrophil infiltration [159]. In piglets healthy colon, luminal fecal transplant increased the relative abundance of obligate anaerobes, mucosal resistance to bacterial adhesion, preserved goblet cell mucin stores and reduced the incidence of induced NEC [277]. Similarly, in murine NEC, FMT prevented tissue injury, decreased intestinal inflammation, improved barrier function via increasing claudin-7 (a tight junction protein), and improved NEC symptoms through reduced free oxygen radicals and increased nitric oxide [276]. Further, FMT reduced TLR4-mediated pro-inflammatory signaling and inflammatory cytokine abundance, and suppressed intestinal apoptosis [274], which decreased bacterial translocation across the intestinal barrier and increased the total intestinal bacterial number [274]. However, rectal FMT administration in preterm piglets following neomycin and amoxicillin-clavulanate antibiotic cessation conferred no protection against NEC, despite FMT administration or antibiotic treatment alone reducing NEC incidence [275]. Antibiotics may induce changes in the gut (altered pH, nutrient availability, immunoreactivity) or overgrowth of antibiotic-resistant species which hinder the beneficial effect of FMT-delivered species.
Despite FMT showing promise in animal NEC models, its translation to humans faces challenges. Given the differences between the adult and the neonatal intestinal microbiome, adult-sourced FMT is potentially unsafe for infants [278]. Sourcing healthy infant donors is extremely challenging, among low gestational age preterm infants athigh risk of NEC. Artificial FMT comprised of laboratory-produced microbial communities with known beneficial species or the manipulation of fecal material such as FFT, that has shown efficacy equal to FMT in piglets [279], may represent more clinically acceptable approaches in NEC. However, significant microbiome variation between individuals further complicates the identification of an ‘ideal’ microbiome for FMT or FFT and the efficacy of combination therapies needs careful evaluation.
Conclusion
NEC is a devastating multifactorial disease that clouds the prognosis of preterm infants. Despite years of research, no gene or group of genes could be singled out as causative, and the variability of the pathogenetic processes leading to NEC and of its clinical course suggests that NEC may not be a single disease but rather a syndrome. The timing of bacterial colonization of the gastrointestinal tract also appears to be a critical factor in NEC development, with certain microbes interacting with the immune system differently to promote either inflammation or tolerance at different times of immune maturation.
As a result, gathering knowledge on precipitating factors and their interactions in driving NEC pathology is highly challenging, which in turn renders devising appropriate diagnostic tools (including biomarkers), developing preventive strategies and safe and effective treatments difficult. However, recent research has made substantial progress in understanding the intestinal dysbiosis and its interplay with the developing immune system in NEC, highlighting the translational promise of immune- and microbiota-based therapeutics. Unravelling the mechanisms of how intestinal bacterial colonization influences the developing immune system of preterm infants, and vice versa, could be the missing link that researchers have been seeking for years in their efforts to combat NEC and ameliorate the immense suffering wrought by this insidious disease.