1H, 13C and 15N backbone resonance assignment of the lytic polysaccharide monooxygenase LsAA9A from Lentinus similis
Piera Wiesinger, Mats Sandgren, Gustav Nestor

TL;DR
This paper reports the backbone resonance assignment of a lytic polysaccharide monooxygenase enzyme using NMR techniques.
Contribution
The study provides a detailed backbone resonance assignment for the apo form of LsAA9A using NMR.
Findings
The 1H, 13C, and 15N backbone resonance assignment of LsAA9A was successfully completed.
The secondary structure prediction using TALOS-N aligns with the crystal structure but shows minor discrepancies.
LsAA9A has a core β-sandwich structure typical of LPMOs.
Abstract
Lytic polysaccharide monooxygenases (LPMOs) are mono-copper binding enzymes involved in the degradation of carbohydrates. The 25 kDa sized LPMO LsAA9A from the basidiomycete Lentinus similis is known to oxidate cellulose and cellooligomers at the C4 position and thus leading to a breakage of the glycosidic bond. LsAA9A has been recombinantly expressed in Escherichia coli with 13C and 15N labelling. Here, we present the 1H, 13C and 15N backbone resonance assignment of the apo form. The secondary structure was predicted using the TALOS-N software and it was overall in agreement with the crystal structure of LsAA9A expressed in E. coli. A few shorter α-helices and β-sheets present in the crystal structure are missing in the NMR prediction and vice versa. LsAA9A resembles the typical structural elements of LPMOs with a core β-sandwich.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
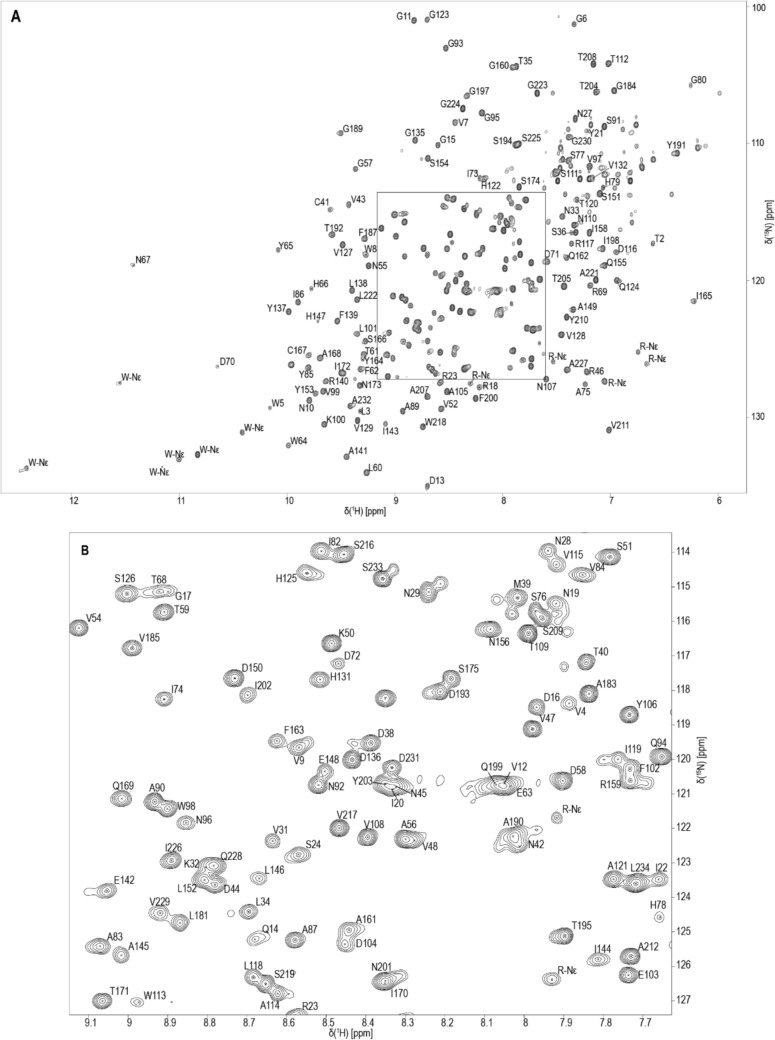 Figure 1
Figure 1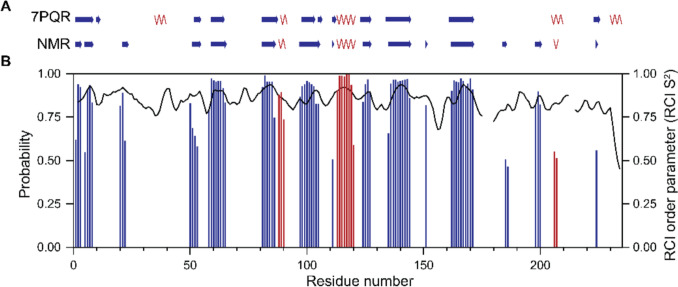 Figure 2
Figure 2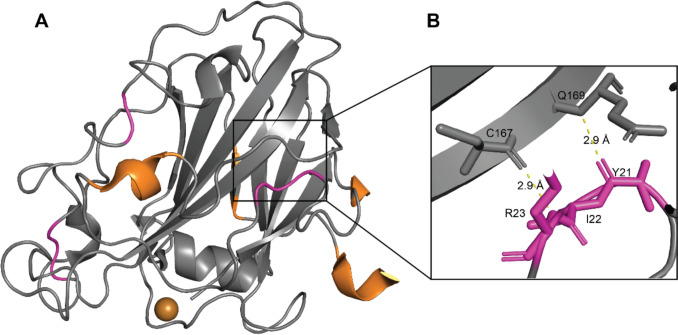 Figure 3
Figure 3- —Swedish University of Agricultural Sciences
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicrobial Natural Products and Biosynthesis · Enzyme Structure and Function · Biofuel production and bioconversion
Biological context
Lytic polysaccharide monooxygenases (LPMOs) are carbohydrate-degrading enzymes predominantly found in fungi and bacteria, but also in viruses, plants and insects (Vandhana et al. 2022). LPMOs cleave glycosidic bonds in an oxidative manner via a mono-copper coordinated by a histidine brace in the active site (Bissaro and Eijsink 2023). The common structure of LPMOs is a core β-sheet sandwich and a few short α-helices connected via loops. LPMOs have a characteristic flat substrate-interaction surface, which also includes the catalytic site. This feature enables LPMOs to act on crystalline polysaccharides such as cellulose or chitin (Aachmann et al. 2012), but they can also be active on shorter poly- and oligosaccharides (Frandsen et al. 2016; Rieder et al. 2021). The described features are crucial for the ability of LPMOs to degrade carbohydrates both for industrial applications as well as in their biological context. Since the first crystal structures of LPMOs (Karkehabadi et al. 2008; Vaaje-Kolstad et al. 2005b) and their boosting effect on the degradation efficiency of cellulose (Harris et al. 2010) and chitin (Vaaje-Kolstad et al. 2005a, ) was revealed about 20 years ago, much research has been focused on industrial applications. More recently, it has been shown that LPMOs play biological roles in pathogenesis, cell wall remodelling and copper transport (Vandhana et al. 2022).
The LPMO LsAA9A from the basidiomycete Lentinus similis has been characterized in depth both biochemically and structurally (Frandsen et al. 2016; Simmons et al. 2017; Tandrup et al. 2023). It is capable of oxidizing cellulose and cellooligomers at the C4 position and other polysaccharides at both the C1 and C4 position (Simmons et al. 2017). The crystal structure of LsAA9A was first solved by Frandsen et al. (2016), who also presented complex structures with cellooligomers bound. These structures have been used as the basis for mechanistic studies such as investigations on the copper coordination sphere upon reduction (Tandrup et al. 2022) or computational studies on the electronic effect of priming reduction, substrate and co-substrate binding (Wieduwilt et al. 2024).
Five catalytic domains of LPMOs, including three bacterial (Aachmann et al. 2011; Christensen et al. 2021; Courtade et al. 2015) and two fungal (Courtade et al. 2016; Kitaoku et al. 2018) LPMOs, have been expressed with isotopic labelling for NMR studies and their backbone resonance assignments have been deposited in BMRB to date. However, no data has been deposited for LsAA9A so far.
In this study, we present the recombinant expression and purification of ^13^C and ^15^N labelled LsAA9A and a nearly complete backbone resonance assignment of apo-LsAA9A. Four residues which are located in a loop in the crystal structure could not be assigned, probably due to intermediate dynamics, making them invisible on the NMR time scale. Additionally, we present a secondary structure prediction using the TALOS-N software. The secondary elements predicted from the NMR data are to a large extent coherent with the crystallographic data. The crystal structure (PDB code 7PQR) displays two α-helices which are absent in the NMR prediction, whereas the predicted NMR structure contains a few additional short β-strands.
Methods and experiments
Protein expression and purification
LsAA9A was recombinantly expressed by a BL21(DE3) E. coli strain containing the plasmid pLyGo-Ec-6_LsAA9A (Hernández-Rollán et al. 2021; Tandrup et al. 2022). The expression was done in M9 minimal media for isotopic labelling purposes using 1 g/l NH_4_Cl (99% ^15^N from Cortecnet, Les Ulis, France) and 2 g/l D-glucose (99% U-^13^C from Cambrige Isotope Laboratories, Inc, Andover, MA, USA). A 5 ml pre-culture in LB medium was incubated for 6 h at 37 °C shaking at 250 rpm. This pre-culture was transferred to 50 ml M9 medium incubating at the same conditions for ~ 14 h. For the protein expression 1 l of M9 minimal medium was inoculated with the 50 ml culture and grown until the OD_600_ was ~ 0.8 at 37 °C with shaking at 180 rpm. The induction was done with 1 mM (final concentration) isopropyl β-d-1-thiogalactopyranoside (IPTG) and the protein was expressed for ~ 20 h at 16 °C shaking at 150 rpm. The cells were harvested by centrifugation (4000 rpm, 20 min). The cell pellet was re-suspended in 35 ml 25 mM Bis–Tris buffer pH 5.9 and stored at −20 °C until further usage.
For protein extraction the suspended cells were incubated with DNase (10 µg/ml) for 1 h gently shaking on ice before and after cell disruption. The cells were disrupted with a Cell Disruptor (Constant Systems Ltd, Daventry, United Kingdom) at 20 kPsi and centrifuged at 18,000 rpm for 40 min. The supernatant was filtered with a 0.45 µm filter.
For purification of LsAA9A, an ÄKTApurifier system and columns from Cytiva (Uppsala, Sweden) were used. The protein extract was desalted with a HiPrep 26/10 Desalting column using a 25 mM Bis–Tris buffer pH 5.9. This step was followed by anion-exchange chromatography purification with 2 × 5 ml HiTrap Q HP columns. The running buffers used were: (A) 25 mM Bis–Tris, pH 5.9 and (B) 25 mM Bis–Tris, 1 M NaCl, pH 5.9. The following gradient was used with a flow rate of 3 ml/min: 0–70 ml sample loading with buffer A, 150 ml linear gradient to 50% buffer B, 50 ml linear gradient to 100% buffer B, 50 ml buffer B, 60 ml buffer A for column regeneration. Fractions containing LsAA9A were pooled, concentrated with a Vivaspin 5 MWCO centrifugal concentrator and filtered with a 0.25 µm filter before loading on a size exclusion column (SEC) (Superdex 75 16/600). The SEC running buffer used was 40 mM NaOAc, 100 mM NaCl, pH 5.9. All buffers were filtered and degassed with 0.45 µm filters before use. Fractions from the SEC purification were analysed by SDS-PAGE for purity determination. Protein concentrations were determined spectrophotometrically at 280 nm with an extinction coefficient of 48,025 M^−1^ cm^−1^.
For the NMR sample the purest LsAA9A fractions were combined and concentrated to 300 µM. The buffer was exchanged to 40 mM sodium phosphate, 10 mM NaCl, pH 6. Before transferring the sample to a Shigemi NMR tube 0.02% NaN_3_ and 10% D_2_O were added, as well as 0.5 mM DSS for chemical shift referencing.
NMR spectroscopy
All NMR experiments were performed at the Swedish NMR centre in Gothenburg on a 900 MHz spectrometer (Oxford 900 magnet with Bruker Avance HDIII console) equipped with a 3 mm TCI ^1^H/^13^C/^15^N CryoProbe. For ^1^H, ^13^C and ^15^N backbone resonance assignment data was collected with the following experiments: 1D 1H, 2D ^1^H,^15^N-HSQC and 3D HNCO, HN(CA)CO, HNCA, HN(CO)CA, HNCACB, CBCA(CO)NH and HBHA(CO)NH. All experiments are standard experiments in the Bruker library. The spectra were recorded at 37 °C with 20–25% non-uniform sampling (NUS) and reconstructed and processed with TopSpin 4.1.4. ^1^H chemical shifts were referenced to DSS (δ = 0.00 ppm) and ^13^C and ^15^N chemical shifts were indirectly referenced according to Wishart et al. (1995). The assignment was performed manually with CcpNmr AnalysisAssign Version 3 (Skinner et al. 2016) and with the input of NMRtist (Klukowski et al. 2022, 2023) chemical shift results (NMRtist calculations performed on 2024–02–22). Predictions from the SHIFTX2 software (Han et al. 2011) were used for assistance.
Secondary structure prediction
Secondary structure elements and the random coil index order parameter S^2^ (RCI) (Berjanskii & Wishart 2005, 2008) were predicted with the TALOS-N program (Shen & Bax 2013). The web-based version of the software was used (https://spin.niddk.nih.gov/bax-apps/nmrserver/talosn/). The predictions are based on backbone resonance chemical shift assignment data including also Hα. It was compared to the crystal structure of LsAA9A expressed in E. coli (PDB: 7PQR) (Tandrup et al. 2022).
Extent of assignment and data deposition
Backbone resonance assignment of LsAA9A
We present the assignment of backbone resonances of the copper-free form (apo-form) of AA9A from Lentinus similis. In earlier experiments, the binding of copper by addition of copper acetate and removing of copper by EDTA was tested. These experiments confirmed that LsAA9A is predominantly without copper after expression in E. coli and purification. The well dispersed and sharp cross-peaks in the 2D ^1^H,^15^N-HSQC indicate a folded enzyme (Fig. 1) which is stable even after several days of data collection at 37 °C. The protein sequence contains 235 amino acid residues of which 20 are prolines. The backbone assignment is to > 84% complete (^1^HN 97%, ^15^N 89%, ^13^Cα 88%, ^13^Cβ 86%, ^13^CO 84% and ^1^Hα 87%). For most prolines the C´, Cα, Cβ and Hα could be assigned. The backbone resonances of six other residues could not be assigned. This includes A133, G220 and residues D176, D177, S178 and T179. G220 is predicted by the SHIFTX2 software to have its HN proton chemical shift near the water signal (~ 5 ppm), which is most likely the reason why it could not be detected. Residue 133 and 176 to 179 are located in loops in the crystal structure (7PQR) in close proximity to each other. We therefore suspect that they are flexible in a dynamic range where a signal could not be detected by NMR spectroscopy. The cross-peaks which we expect to correspond to side-chain NH resonances of six tryptophan and seven arginine residues have been marked as W/R-Nε respectively (Fig. 1). The chemical shift data including N, HN, C´, Cα, Cβ, Hα and Hβ has been deposited at the Biological Magnetic Resonance Bank (BMRB), with the accession code 53342.Fig. 1^1^H,^15^N-HSQC spectrum of LsAA9A (A) with an expansion of the middle region (B) shown below. The spectrum was acquired from a sample of 300 µM ^13^C,^15^N-LsAA9A in 40 mM sodium phosphate, 10 mM NaCl, pH 6 and 10% D_2_O at 37 °C on a 900 MHz spectrometer
Secondary structure of LsAA9A in solution
The protein backbone torsion angles and secondary structure of LsAA9A in solution were predicted by the TALOS-N software package. In Fig. 2 the structural elements in the crystal structure (PDB code: 7PQR) and NMR prediction, as well as the probability of a predicted structural element and the random coil index order parameter S^2^ are shown.Fig. 2. Secondary structure elements of apo-LsAA9A predicted by TALOS-N: (A) schematic comparison of α-helices (red) and β-sheets (blue) observed in the crystal structure (7PQR) and predicted from NMR backbone resonance assignment data, (B) probability of structural elements (α-helices (red bars) and β-sheets (blue bars)) and random coil index (RCI) order parameter S^2^ (black line) for each residue
Overall, the structural elements of LsAA9A are coherent between NMR data and the X-ray crystal structure. Some minor differences are observed in the length or exact start of α-helices or β-sheets. However, also some major differences were observed and visualized in Fig. 3. Additional to the NMR prediction, the X-ray structure contains a short β-strand at position 12 and 13 as being antiparallel to the previous β-sheets. In contrast, the NMR prediction contains a β-strand at position 21 to 23, which is absent in the X-ray structure. The N(H)-CO distance to the neighbouring major β-sheet in the crystal structure is 2.9 Å. This distance is also observed between other β-sheets in the enzyme. We therefore conclude that the residues 21 to 23 can form a short β-strand interacting with residues 167–169. The crystal structure shows a helix from position 36 to 40, which is not predicted from the NMR data. At position 186/187 and 199–201 TALOS-N predicts two β-strands, which are absent in the crystal structure. Both the following α-helix in the crystal structure at position 206–210 and β-strand at 224–226 are much shorter in the NMR secondary structure prediction. The very last α-helix from position 231 to 235 is absent in the NMR structure and marked as dynamic by TALOS-N. Some of the predictions, such as residues 186/187 and 224–226, have a probability of just above the TALOS-N threshold of 0.5. Therefore, the prediction should be taken with caution.Fig. 3. Crystal structure (7PQR) with highlighted differences compared to NMR secondary structure prediction. The copper ion (gold) is present in the LsAA9A crystals, but not in the NMR sample. (A) α-helices and β-strands observed in the X-ray crystal structure, but not in the NMR structure prediction are marked in orange. Loops in the crystal structure predicted as β-strands from the NMR data are marked in magenta. (B) Zoom-in on potential β-strand formation of residues 21–23 and interaction with the major β-sheet, specifically hydrogen bonding (N(H)-CO 2.9 Å) to residues 167–169
All differences of the secondary elements between NMR data and X-ray crystal structure are located outside the core β-sandwich and outside the substrate binding site. The distance to the copper binding site is at least 15 Å. It should be pointed out that the NMR data was collected on the apo-form (copper free) protein and X-ray data on the holo-form (copper bound). However, due to the lack of proximity to the copper we believe that the differences in secondary structure cannot be related to the absence or presence of copper, but rather to the protein being in a dynamic or static environment.