Genome‐Driven Analysis Reveals the Biotechnological Potential of a Novel Paenibacillus sp. Isolated From Crude Oil
João Victor dos Anjos Almeida, Carlos Miguel Nóbrega Mendonça, Leandro Marcio Moreira, Ricardo Pinheiro Pinheiro de Souza Oliveira, Alessandro de Mello Varani, Mauro de Medeiros Oliveira

TL;DR
A new Paenibacillus strain from crude oil has a genome that suggests it can help with biofuel production, bioremediation, and agriculture.
Contribution
The study presents the complete genome of a novel Paenibacillus sp. strain with biotechnological potential revealed through in silico analysis.
Findings
The genome contains 259 CAZyme genes, indicating strong polysaccharide-degrading capabilities for biofuel production.
Complete B-vitamin biosynthesis pathways and antimicrobial biosynthetic gene clusters suggest metabolic autonomy and biocontrol potential.
Incomplete pathways for B2 and K2 vitamins imply possible syntrophic interactions with other microbes.
Abstract
Microbial biotechnology plays a critical role in addressing environmental challenges and promoting sustainability. Here, we report the complete genome sequencing of Paenibacillus sp. strain 210, previously isolated from Brazilian crude oil and known for its levan metabolism and biosurfactant production. With the sequenced genome, we employed bioinformatics tools for assembly and annotation, followed by comprehensive in silico analyses, including phylogenomics, biosynthetic gene cluster (BGC) identification, carbohydrate‐active enzyme (CAZyme) profiling, and metabolic pathway reconstruction. The assembled 5.7 Mb genome harbors four prophage regions and 13 antimicrobial BGCs, including those encoding fusaricidin, paenicidin A, paenilan, paeninodin, and tridecaptin. Phylogenomic analysis combined with average nucleotide identity measurements indicates that this strain does not cluster with…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
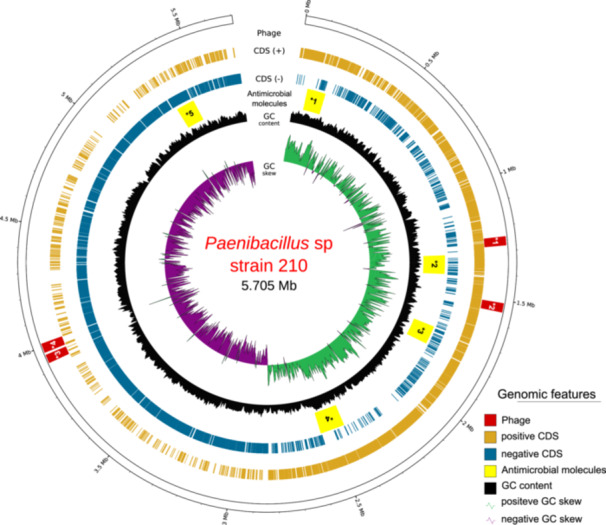 Figure 1
Figure 1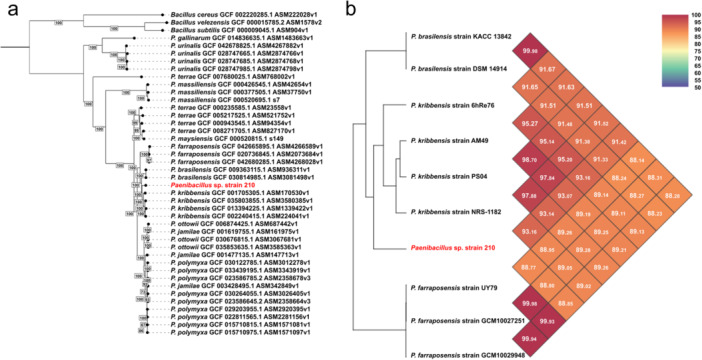 Figure 2
Figure 2 Figure 3
Figure 3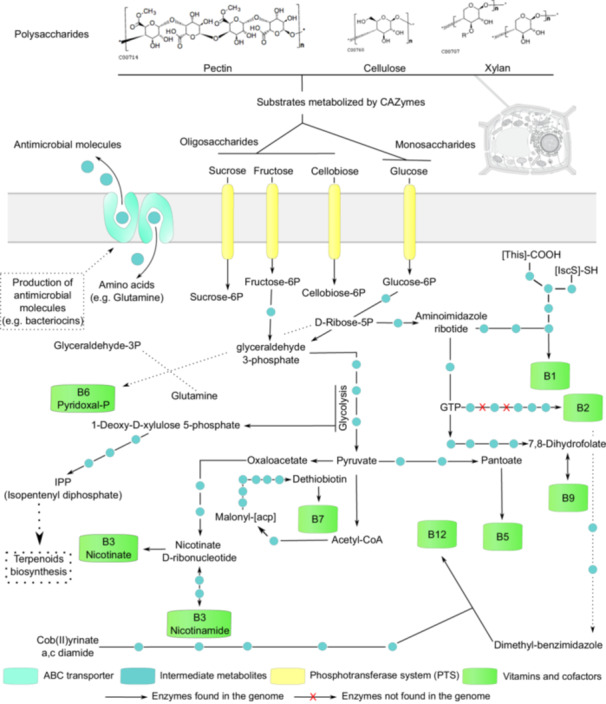 Figure 4
Figure 4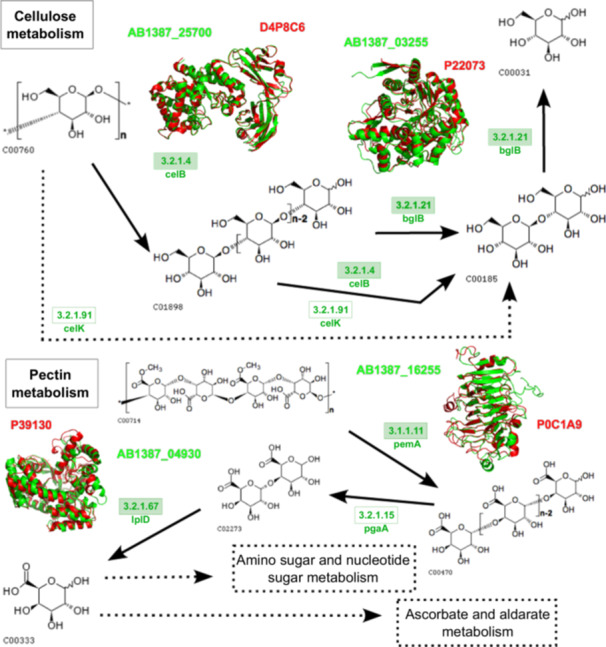 Figure 5
Figure 5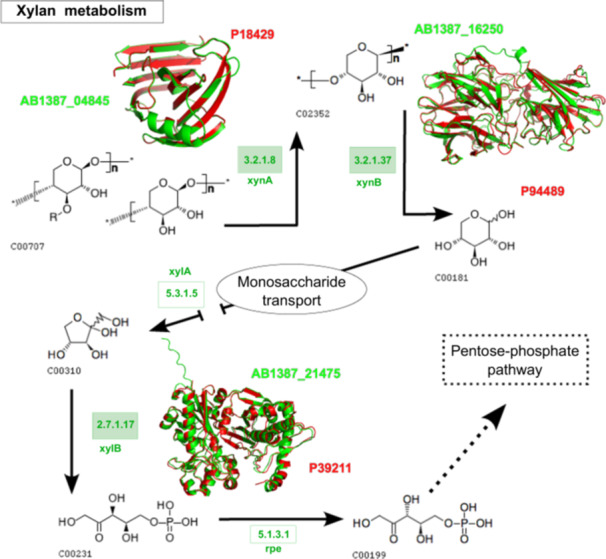 Figure 6
Figure 6| Features |
|
|---|---|
| Genomic size (bp) | 5,705,131 |
| Sequencing depth (mean) | 86× |
| GC content (%) | 46.5 |
| Genes (total) | 5170 |
| CDSs (total) | 5040 |
| tRNA | 92 |
| 5S rRNA | 11 |
| 16S rRNA | 11 |
| 23S rRNA | 11 |
| ncRNA | 4 |
| Pseudogenes | 123 |
| CRISPR arrays | 9 |
| Antibiotic resistance‐related genes (ABRICATE) | ‐ 1× (AGly)aadE‐Pp (aminoglycoside) |
| ‐ 1× (Rif)rphD (rifamycin) | |
| Virulence‐related genes |
Hemolysin III family protein 3x Hemolysin family protein |
| CheckM (completeness/contamination) | 99.72/1.25 |
| BUSCOs (completeness) | 99.3 |
| Features | Psp | Pbr | Pkr | Ppe | Pfa | Pot | Ppo | Pja | Pte | Pma |
|---|---|---|---|---|---|---|---|---|---|---|
| Country | Brazil | South Korea | South Korea | South Korea | Uruguay | EUA | China | India | South Korea | China |
| Isolation source | Crude oil | Soil | Soil | Soil | Nodule endophyte | anaerobic digestate | Soil | Rice seed | Soil | Corn rhizosphere |
| Plasmid | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| AMR | ||||||||||
| (Agly)aadE‐Pp | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 |
| clbB | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| RPH | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 |
| Prophage | ||||||||||
|
| quest | 0 | inc | 0 | 0 | quest + int | int | 0 | 0 | |
|
| quest | quest | int | quest | quest | quest | quest | 0 | quest | quest |
|
| int | 0 | int | int | int | 0 | 0 | 0 | int | int |
|
| quest | 0 | 0 | quest | 0 | 0 | 0 | 0 | 0 | int |
|
| 0 | int | 0 | 0 | int | 0 | 0 | 0 | 0 | 0 |
| Deep‐sea thermophilic phage D6E ( | 0 | quest | 0 | 0 | 0 | 0 | 0 | 0 | quest | 0 |
|
| 0 | int | 0 | 0 | inc | 0 | inc | 0 | 0 | 0 |
|
| 0 | inc | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
|
| 0 | quest | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Bacteriophage lily ( | 0 | 0 | 0 | 0 | int | 0 | 0 | 0 | 0 | 0 |
|
| 0 | 0 | 0 | 0 | inc | 0 | 0 | 0 | 0 | 0 |
|
| 0 | 0 | 0 | 0 | inc | 0 | 0 | 0 | 0 | 0 |
|
| 0 | 0 | 0 | 0 | inc | 0 | 0 | 0 | 0 | 0 |
|
| 0 | 0 | 0 | 0 | inc | 0 | 0 | 0 | 0 | 0 |
|
| 0 | 0 | 0 | 0 | 0 | 2x_quest | 0 | 0 | 0 | inc |
|
| 0 | 0 | 0 | 0 | 0 | 0 | int | 0 | 0 | 0 |
|
| 0 | 0 | 0 | 0 | 0 | 0 | int | 0 | 0 | 0 |
|
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | quest | quest | 0 |
|
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | quest | 0 | 0 |
|
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | int |
|
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | inc |
| Gene | Product | Role in IAA biosynthesis | Gene ID |
|---|---|---|---|
|
| Chorismate synthase | Converts 5‐enolpyruvylshikimate‐3‐phosphate to chorismate | AB1387_14830 |
|
| Chorismate mutase | Catalyzes the conversion of chorismate to prephenate | AB1387_14820 |
|
| 3‐Deoxy‐7‐phosphoheptulonate synthase | Key enzyme in the shikimate pathway | AB1387_07710 |
|
| Tryptophan synthase subunit alpha | Converts indole‐3‐glycerol phosphate to tryptophan | AB1387_14790 |
|
| Tryptophan synthase subunit beta | Converts indole‐3‐glycerol phosphate to tryptophan | AB1387_14795 |
|
| Tryptophan‐transfer RNA ligase | Activates tryptophan for protein synthesis | AB1387_20675 |
|
| Indole‐3‐glycerol phosphate synthase | Catalyzes indole‐3‐glycerol phosphate synthesis | AB1387_14805 |
|
| Anthranilate phosphoribosyltransferase | Converts anthranilate to phosphoribosylanthranilate | AB1387_14810 |
|
| Anthranilate synthase component I | Synthesizes anthranilate from chorismate | AB1387_14815 |
|
| Glutamine amidotransferase | Supports anthranilate synthase activity | AB1387_18125 |
|
| Phosphoribosylanthranilate isomerase | Converts phosphoribosylanthranilate to carboxyphenylaminodeoxyribulose phosphate | AB1387_14800 |
|
| Thiamine pyrophosphate‐binding protein | Involved in indole‐3‐pyruvate decarboxylation | AB1387_00400 |
| Gene | Product | Role in phosphate solubilization | Gene ID |
|---|---|---|---|
|
| Phosphatase PAP2 family protein | Hydrolyzes organic phosphate esters | AB1387_21265 |
|
| Aminopeptidase | Degrades organic phosphate compounds | AB1387_11915 |
|
| Alkaline phosphatase | Releases inorganic phosphate from organic sources | AB1387_21945 |
|
| Phosphonate ABC transporter permease | Transports phosphonates | AB1387_21795 |
|
| Phosphonate ABC transporter substrate‐binding protein | Binds extracellular phosphonates | AB1387_21780 |
|
| Phosphonate ABC transporter ATP‐binding protein | Energizes phosphonate transport | AB1387_21785 |
|
| Phosphate ABC transporter substrate‐binding protein | Binds extracellular phosphate | AB1387_08280 |
|
| Phosphate ABC transporter permease subunit | Transports phosphate across the membrane | AB1387_08285 |
|
| Phosphate ABC transporter permease PstA | Transports phosphate across the membrane | AB1387_08290 |
|
| Phosphate ABC transporter ATP‐binding protein | Energizes phosphate transport | AB1387_08295 |
|
| Phosphate signaling complex protein | Regulates phosphate uptake | AB1387_08370 |
| Gene | Product | Role in nitrogen metabolism | Gene ID |
|---|---|---|---|
|
| Respiratory nitrate reductase subunit gamma | Reduces nitrate to nitrite | AB1387_08500 |
|
| Nitrate reductase molybdenum cofactor assembly chaperone | Assists in nitrate reductase assembly | AB1387_08495 |
|
| Nitrate reductase subunit beta | Catalyzes nitrate reduction | AB1387_08490 |
|
| Nitrate reductase subunit alpha | Catalyzes nitrate reduction | AB1387_08485 |
|
| Nitrate/nitrite transporter | Transports nitrate/nitrite across membranes | AB1387_08465 |
|
| Nitrite reductase small subunit | Reduces nitrite to ammonia | AB1387_03300 |
|
| Nitrate/nitrite transporter | Transports nitrite | AB1387_16035 |
|
| Nitrite reductase large subunit | Reduces nitrite to ammonia | AB1387_03295 |
|
| Ammonium transporter | Imports ammonium into the cell | AB1387_08985 |
| Gene | Product | Role in nitrogen fixation | Gene ID |
|---|---|---|---|
|
| Nitrogenase iron protein | Electron donor for nitrogenase | AB1387_05115 |
|
| Nitrogenase cofactor biosynthesis protein | Assembles FeMo‐cofactor | AB1387_05135 |
|
| Nitrogenase cofactor biosynthesis protein | Synthesizes FeMo‐cofactor precursor | AB1387_05110 |
|
| Nitrogenase molybdenum–iron protein alpha chain | Catalyzes N₂ reduction | AB1387_05120 |
|
| Nitrogenase Fe─S cluster assembly protein | Assists in Fe─S cluster formation | AB1387_20555 |
|
| Nitrogenase cofactor biosynthesis protein | Assembles FeMo‐cofactor | AB1387_05130 |
|
| Nitrogenase molybdenum–iron protein subunit beta | Catalyzes N₂ reduction | AB1387_05125 |
|
| Nitrogen fixation protein | Binds FeMo‐cofactor intermediates | AB1387_05140 |
|
| HesA/MoeB/ThiF family protein | Involved in molybdopterin biosynthesis | AB1387_05145 |
- —This project was financed by the São Paulo Research Foundation (FAPESP; Project Number 2018/25511‐1; 2020/13271‐6 [C.M.N.M]; 2023/04372‐1 [M.M.O.]). João Victor dos Anjos Almeida was supported by a CA
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAlgal biology and biofuel production · Microbial Metabolism and Applications · Biofuel production and bioconversion
Introduction
1
Microbial biotechnology plays a crucial role in addressing urgent global challenges, such as environmental degradation, food security, and public health. These areas form essential pillars in sustainability agendas, aligning with the objectives established in the United Nations' 2030 Agenda. Microorganisms, renowned for their remarkable metabolic diversity and resilience in extreme environments, are thus integral to developing sustainable solutions.
Within this context, certain microbial species play key roles in decomposing complex organic materials. By recycling biological resources, they propel nutrient cycles and help stabilize microbial communities. Polysaccharides, structurally intricate carbon sources, are central to recycling processes across diverse ecosystems—ranging from the human gut to marine environments (Sichert and Cordero 2021). Their degradation yields products that nourish other microorganisms, consequently boosting microbial diversity and functionality (Sichert and Cordero 2021).
Among these polysaccharides, those derived from plants—such as pectin, hemicellulose, and cellulose—constitute critical components of plant cell walls and are abundantly present in agricultural and forestry residues. When efficiently broken down, these polysaccharides produce fermentable sugars vital for bioethanol production (A. P. De Souza et al. 2013). While their complete hydrolysis poses technical challenges, it also offers notable opportunities for advancing sustainable biofuel technologies, waste valorization, and reducing reliance on petroleum‐based energy sources (A. P. De Souza et al. 2013; Zabed et al. 2017).
Enzymatic hydrolysis, employing specialized enzymes (e.g., cellulases and hemicellulases, such as xylanases) to cleave glycosidic bonds, followed by microbial fermentation (Zabed et al. 2017), can transform these biomasses into renewable energy. For example, nonstructural carbohydrates in sugarcane straw degrade gradually, whereas structural carbohydrates often persist (Pagliuso et al. 2021), complicating their utilization. Metabolizing these remaining structural carbohydrates is therefore key to second‐generation (2G) ethanol production (A. P. De Souza et al. 2014), a pursuit that gains urgency given projections that energy crops could meet one‐third of global energy needs by 2050 (Guo et al. 2015).
Moreover, certain microbial species can produce and release essential vitamins in polluted environments, thereby stabilizing microbial communities and supporting the remediation of areas contaminated by oil spills and related pollutants (Babalola 2010; Jonsson and Östberg 2011; Uebanso et al. 2020; Radice et al. 2023). In aquatic settings, these vitamins promote the growth of microalgae used in bioremediation efforts (Radice et al. 2023) or stimulate other microorganisms capable of degrading oil residues in soil (Jonsson and Östberg 2011), aiding ecosystem recovery. Their benefits extend beyond microbial communities to plants (Babalola 2010) and animals (Uebanso et al. 2020), yielding cascading positive effects throughout the ecosystem.
Consequently, microorganisms present innovative solutions to renewable energy challenges, notably by degrading polysaccharides and generating bio‐based compounds suitable as sustainable energy sources. Within this context, a strain of Paenibacillus isolated from Brazilian crude oil samples demonstrated in vitro potential for levan metabolism, as well as the production of biosurfactants and bioemulsifiers (Mendonça et al. 2021). These traits suggest promising applications in enhanced oil recovery, accelerated bioremediation via improved hydrocarbon degradation, and the development of biofuels through the conversion of plant polysaccharides into fermentable sugars (Jonsson and Östberg 2011; A. P. De Souza et al. 2014; Mendonça et al. 2021).
Paenibacillus species occupy diverse habitats—from soils and plant rhizospheres to extreme environments, such as polar regions and deserts—and exhibit multifunctional traits relevant to agriculture, medicine, and biotechnology (Grady et al. 2016). Originally classified within Bacillus based on morphology and endospore formation, Paenibacillus with genome sizes ranging from 3.02 to 8.82 Mbp, gene counts from 3064 to 8478, and G + C content between 39% and 59% (Grady et al. 2016).
Species within the genus Paenibacillus have emerged as versatile biocontrol agents, offering sustainable solutions against antimicrobial resistance (AMR) and enhancing agricultural productivity. They produce antifungal lipopeptides (e.g., fusaricidin and paenimyxin), hydrolytic enzymes (e.g., chitinases and glucanases), and volatile organic compounds that suppress plant pathogens such as Fusarium oxysporum, Botrytis cinerea, and Rhizoctonia solani (Jeong et al. 2019; Yuan et al. 2022; Dobrzyński and Naziębło 2024). Commercial formulations exploit these traits; for example, products derived from Paenibacillus polymyxa control fungal diseases in peppers and strawberries via fusaricidins and polymyxins (S. H. Lee et al. 2013; Jeong et al. 2019; Tsai et al. 2022), while Paenibacillus elgii JCK1400, producing pelgipeptins, targets tomato gray mold and wheat rust (Kim et al. 2020). Paenibacillus spp. also promote plant growth through indole‐3‐acetic acid (IAA) production, phosphate solubilization, and nitrogen fixation, enhancing soil fertility and crop resilience (Jeong et al. 2019; Do Carmo Dias et al. 2021; Yuan et al. 2022). For instance, Paenibacillus peoriae ZBSF16 harbors ipdC and nif clusters for IAA biosynthesis and nitrogen fixation, while producing fusaricidins, exemplifying its dual agroecological potential (Yuan et al. 2022). Beyond agriculture, species such as P. peoriae and Paenibacillus validus inhibit clinically relevant pathogens, including Listeria monocytogenes, Candida spp., and Staphylococcus aureus, highlighting their broader biotechnological relevance (Lorentz et al. 2006).
Building upon previous characterization of Paenibacillus sp. strain 210 (Mendonça et al. 2021), we used comparative genomics to explore the genetic traits underlying its potential for various biotechnological applications. In this study, we present the complete genome sequence of strain 210 and analyze its genetic determinants of metabolic versatility. Our findings reveal a robust capacity for degrading complex polysaccharides—namely, cellulose, pectin, and xylan—along with complete biosynthetic pathways for B vitamins and genes linked to plant growth promotion and pathogen biocontrol. Collectively, these results underscore the strain's potential for diverse biotechnological applications, most notably in bioethanol production, bioremediation, and plant growth promotion.
Materials and Methods
2
Genomic Sequencing of the Isolate
2.1
Paenibacillus sp. strain 210, isolated from heavy crude oil samples, was collected from a Brazilian oil well in the Potiguar Basin—for further details, see Mendonça et al. (2021)—and stored in the Microbial Biomolecules Laboratory (LBM) collection at the University of São Paulo.
For cultivation, Paenibacillus sp. strain 210 was grown on de Man, Rogosa e Sharpe (MRS) agar under microaerophilic conditions at 37°C for 12–14 h, and single colonies were subsequently propagated in liquid MRS medium under the same conditions. The harvested cell pellets were preserved in 20% (v/v) glycerol at −20°C. Genomic DNA was then extracted using the PureLink Genomic DNA Mini Kit (Invitrogen, Thermo Fisher Scientific) according to the manufacturer's instructions, with DNA quality and concentration assessed via spectrophotometry and fluorometry. Sequencing libraries were prepared following the PacBio HiFi workflow, and the genome was sequenced at the Arizona Genomics Institute using the PacBio HiFi REVIO platform.
Genome Assembly and Annotation
2.2
The generated reads were assembled with hifiasm v0.19.8 (Feng et al. 2022). Following assembly, the genome was oriented using DNAapler (Bouras et al. 2024), beginning at the dnaA gene. Genome quality was assessed with CheckM v1.2.1 (Parks et al. 2015). Putative plasmid sequences were subsequently evaluated using Deeplasmid (Andreopoulos et al. 2022) and PLASMe (Tang et al. 2023), with results cross‐validated against publicly available reference genomes.
Genome annotation was conducted using the National Center for Biotechnology information (NCBI) Prokaryotic Genome Annotation Pipeline (PGAP) v2024‐04‐27 (Tatusova et al. 2016). To refine these annotations, we employed eggNOG‐Mapper v2.1.1 (Cantalapiedra et al. 2021) to map PGAP‐annotated genes to known gene families and InterProScan v5.67‐99.031 (Jones et al. 2014) to predict protein domains, comparing the results with reference genes from corresponding metabolic pathways. Prophage regions were identified using PHASTEST (Wishart et al. 2023), and circular genome representations were generated with the GenoVi pipeline v0.4.3 (Cumsille et al. 2023).
To detect antibiotic resistance genes (ARGs), protein‐coding nucleotide sequences were compared against databases, including NCBI AMRFinderPlus (Feldgarden et al. 2021), CARD (Alcock et al. 2023), ResFinder (Bortolaia et al. 2020), ARG‐ANNOT (Gupta et al. 2014), MEGARES (Doster et al. 2020), and PlasmidFinder (Carattoli et al. 2014) using anti‐biotic resistance screening tool in contigs for antimicrobial resistance or virulence genes (ABRICATE) v1.0.1 (https://github.com/tseemann/abricate). Biosynthetic gene clusters (BGCs) associated with antimicrobial activity were identified using BAGEL v4.0 (van Heel et al. 2018) and antiSMASH v7.1 (Blin et al. 2023), enabling the detection of additional regions linked to other secondary metabolites.
The genomic neighborhoods of the biosynthetic clusters were manually inspected and visualized using LoVis4u v0.1.4.1 (Egorov and Atkinson 2025).
Annotated protein sequences were analyzed for carbohydrate‐active enzyme (CAZyme)‐related functions by mapping them to the CAZy database (Drula et al. 2022) using dbCAN3 (Jinfang et al. 2023), integrating results from HMMER (Potter et al. 2018), dbCAN‐sub, and DIAMOND (Buchfink et al. 2021) to maximize detection accuracy.
In Silico Taxonomic Assignment Placement and Phylogenomic Analysis
2.3
The taxonomic classification was determined using genome taxonomy database (GTDB)‐Tk v2 (Chaumeil et al. 2022) in conjunction with data from the Genome Taxonomy Database (GTDB, release 214).
To determine the species affiliation of strain 210, we performed a phylogenomic analysis using 253 reference genomes of Paenibacillus species obtained from NCBI data sets. A phylogenomic tree was constructed with IQ‐TREE2 v2.0.7 (Minh et al. 2020) based on a matrix generated by the benchmarking universal single‐copy orthologs (BUSCO) phylogenomics pipeline (https://jamiemcgowan.ie/), using 37 single‐copy core genes (shared by 99.5 of the genomes) identified with BUSCO v5.8.2 (Simão et al. 2015) with the bacillales_odb12 database. As the outgroup, we used Bacillus cereus (GCF_002220285.1), Bacillus subtilis (GCF_000009045.1), and Bacillus velezensis (GCF_000015785.2). The best clustering model, identified by ModelFinder (Kalyaanamoorthy et al. 2017) based on bayesian information criterion, was SYM + I + G4. Branch support was assessed with 1000 bootstrap replicates.
Similarly, we reconstructed the phylogenomic relationships within the clade containing Paenibacillus sp. using additional genomes, supporting its evolutionary placement (308 single‐copy core genes shared by all genomes). For this analysis, the best‐fit model was GTR + F + I + G4.
For taxonomic support, average nucleotide identity (ANI) values were compared among the closest relatives of Paenibacillus sp. We used OrthoANI (I. Lee et al. 2016) for comparisons involving fewer than 10 genomes and ANIclustermap v1.4.0 (https://github.com/moshi4/ANIclustermap) for comparisons involving more than 10 genomes.
Metabolic Pathways Reconstruction
2.4
For the reconstruction of degradation and biosynthesis pathways, KEGG Orthology (KO) terms annotated by eggNOG‐Mapper were referenced against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database and pathways (Kanehisa and Goto 2023).
The metabolic pathways consulted were arabinose (Watanabe et al. 2006; W. R. De Souza 2013), cellulose (KEGG: map00500), fructans (KEGG: ec00051; Buntin et al. 2017), pectin (KEGG: map00040), raffinose (KEGG: map00052), and xylan (KEGG: rn00040 and RAST—Overbeek et al. 2014) for plant polysaccharides, and isopentenyl pyrophosphate (IPP) (Jang et al. 2011), B1/B5/B12/K2 (KEGG: map01240), B2 (KEGG: map00740), B3 (Luo et al. 2023), precursor of B6 (KEGG: rn00750), B7 (KEGG: rn00780), and B9 (KEGG: map00790), for vitamins and precursors.
Molecular Modeling of Candidate Enzymes
2.5
Signal peptides were first identified and removed, if present, using SignalP 6.0 (Teufel et al. 2022) before molecular modeling. The refined sequences were then used for structure prediction with the AlphaFold v3 platform (Abramson et al. 2024), ensuring accurate functional validation of the selected enzymes. We used the SAVES 6.1 platform (https://saves.mbi.ucla.edu/) for structural evaluations, and the models were visualized using PyMOL v3.1.3.1. Additionally, predictions were cross‐referenced with UniProt (https://www.uniprot.org/) to ensure consistency with experimental data. Three‐dimensional structure superposition was calculated using the FATCAT 2.0 tool (Z. Li et al. 2020). All figures were edited with Inkscape v1.4 (https://inkscape.org).
Unless otherwise specified, all software tools utilized were run using their default parameters.
Results
3
Genomic Features and Species‐Level Definition of the Paenibacillus sp. Strain 210
3.1
A total of 928,813 HiFi reads were generated and assembled into a single contig representing a complete circular genome spanning 5.705 Mb, with no detectable plasmids (accession number CP160863). The genome has a guanine and cytosine (GC) content of 46.5% and an average sequencing coverage of 86× (Figure 1).
*Circular representation of the Paenibacillus sp. strain 210 genome (5.705 Mb), where the rings display different genomic features. The outermost ring indicates the identified prophage regions (in the dark red with *1 questionable completeness with NC_03094, *2 intact completeness with NC_028805, *3 questionable completeness with NC_048651, and *4 questionable completeness with NC_048762), followed by the distribution of coding sequences (CDS) on the positive strand (in golden beige) and the negative strand (in blue). The next ring highlights gene clusters associated with the production of antimicrobial molecules (in yellow with *1 fusaricidin B, *2 paeninodin, 3 paenilan, 4 tridecaptin, and 5 paenicidin A). Further inward, the GC content graph (in black) reflects variations in nucleotide composition throughout the genome. At the center, the positive GC skew (in green) and negative GC skew (in purple) are shown.
The genome harbors four prophage regions and five regions associated with antimicrobial molecules production (Figure 1). Annotation with the NCBI PGAP identified a total of 5178 genes (Table 1). Quality assessment by CheckM indicated 99.72% completeness, while BUSCO analysis confirmed 99.30% completeness.
Taxonomic classification using GTDB‐Tk was not achieved, as the ANI values failed to meet the program's threshold. The closest relatives identified included Paenibacillus kribbensis (GCF 002240415.1; 92.91% ANI), P. peoriae (GCF 000236805.1; 92.57% ANI), Paenibacillus brasilensis (GCF 009363115.1; 91.1% ANI), and P. polymyxa (GCF 001719045.1; 87.35% ANI), among others.
This ambiguity was corroborated by complementary BLAST analysis in the NCBI repository using the rpoB (Da Mota et al. 2004) gene sequence—considering the inconclusive classification by the 16S sequence evaluated in a previous study, accession number MW577094 (Mendonça et al. 2021). For that gene, the top hits were P. kribbensis PS04 (GCF 013394225.1; 99% coverage and 96.56% identity), P. brasilensis KACC 13842 (99% coverage and 96.50% identity), P. peoriae KCTC 3673 (GCF 009363115.1; 99% coverage and 96.36% identity), and P. polymyxa HY96‐2 (GCF 002893885.1; 100% coverage and 94.42% identity).
We then reconstructed the phylogenomic relationships between our strain and reference genomes of Paenibacillus species (Figures 2a and Supporting Information Figures A1 and A2 and Table A1), which enabled a more precise identification of its position within the Paenibacillus genus. Together with the ANI cluster data, our analyses indicate that the distance on average genome identities to strain 210 is less than 93% identity, allowing us to classify it as a new species (Figure 2b and Supporting Information Figure A3).
(a) Phylogenetic tree showing the relationships between strain 210 (red) and Paenibacillus species. (b) Heatmap generated by OrthoANI, representing the average nucleotide identity (ANI) values between the genomes of the clade highlighted in green in the phylogeny, with the color gradient indicating the degree of similarity.
Comparing the genomes used in the ANI analysis of the Paenibacillus sp. strain 210 clade, we identified reference species associated with both soil and plants, including endophytic (e.g., Paenibacillus farraposensis) and rhizospheric (e.g., Paenibacillus maysiensis) forms (Table 2). Furthermore, most genomes lacked identifiable plasmids and exhibited distinct patterns in bacteriophage region predictions. The distribution of AMR and phage regions for the other reference genomes can be found in Supporting Information Tables A2 and A3, respectively.
Regarding the shared biosynthetic regions for antimicrobial molecules in Paenibacillus sp., fusaricidin B (Supporting Information Figure A4), paeninodin (Supporting Information Figure A5), and tridecaptin (Supporting Information Figure A6) are conserved across the nine taxonomically related species (Supporting Information Table A4). However, paenilan (Supporting Information Figure A7) is shared with more distant species, according to the phylogenomic relationships we reconstructed, but is absent in the closest genomes. No predictions for paenicidin were found in the closest species clade, except in Paenibacillus sp. (although its characterization has already been observed in P. polymyxa NRRL B‐30509 in literature, Lohans et al. 2012). Additionally, Paenibacillus tianmuensis (GCF_900100345.1) showed predictions for paenicidin but with an incomplete biosynthetic cluster (Supporting Information Figure A8).
CAZyme Profiles of Paenibacillus sp. and Other Paenibacillus sp. Genomes
3.2
Using dbCAN3, we identified a diverse CAZyme repertoire in Paenibacillus sp. strain 210, including glycoside hydrolases (GHs), carbohydrate esterases (CEs), polysaccharide lyases (PLs), carbohydrate‐binding modules (CBMs), and auxiliary activities (AAs). Strain 210 encodes 259 CAZymes (Figure 3), a genomic feature consistent with the genus Paenibacillus, as shown in comparative analyses with reference genomes (Supporting Information Table A5). Substrate specificity predictions linked these enzymes to the degradation of cellulose, xylan, and pectin, corroborated by the abundance of families associated with these polysaccharides in the heatmap (Figure 3).
CAZyme profiles associated with polysaccharide degradation in different Paenibacillus genomes, specifically within the phylogenomic clade most closely related to Paenibacillus sp. (highlighted in red). Heatmaps are provided for: GH (a), CBM (b), GT (c), CE (d), and PL (e). The dendrogram reflects the similarity between CAZyme profiles based on the presence and abundance, calculated using Euclidean distance (top, species relationships; left, CAZyme family relationships). AAs, auxiliary activities; CAZyme, carbohydrate‐active enzyme; CBMs, carbohydrate‐binding modules; CEs, carbohydrate esterases; FOS, fructooligosaccharides; GHs, glycoside hydrolases; GT, glycosyltransferase; PLs, polysaccharide lyases; RFO, raffinose family oligosaccharides.
The GH profile of strain 210 (Figure 3a) revealed GH43 as the most prominent family, with 12 predicted alpha‐arabinofuranosidases and beta‐xylosidases. These enzymes, critical for xylan degradation, were frequently coupled with CBM36 and CBM91 (indicated by adjacent color blocks in Figure 3a). GH32 (green, abundance level 4), associated with fructan metabolism (e.g., inulinases), was also highly represented, with eight entries linked to CBM66 and CBM38 (Figure 3b). Among the compared genomes, strain 210 exhibited the highest GH diversity (55 families), followed by Paenibacillus terrae (52 families), as visualized by the extended teal and green blocks in the heatmap (Figure 3a). In the CEs group, strain 210 did not dominate in CE abundance (Figure 3b), its profile highlighted enzymes targeting acetylxylan (orange gradients, linked to CE1 and CE4), associated with xylanolytic activity. For glycosyltransferases (GTs), strain 210 presented 61 GT families (Figure 3c), comparable to most genomes of the clade. Paenibacillus jamilae presented the highest number of GTs (69 families). Finally, for the PLs group, strain 210 exclusively encoded PL9 and PL10 (purple blocks, Figure 3e), associated with pectate/pectin degradation, highlighting its potential for plant biomass breakdown.
Characterization of Identified Metabolic Pathways
3.3
A total of 16 metabolic pathways were investigated by reconstructing these processes through KO annotations using eggNOG‐mapper. To validate our findings, we compared shared protein domains between reference sequences cataloged in the KEGG database for each pathway. This approach enabled the identification of sequences with common domains, thereby reinforcing the accuracy of the pathway assignments (Supporting Information Table A6).
Through in silico reconstruction of metabolic pathways in our Paenibacillus sp. strain 210, we identified genomic features suggesting a strong capacity to degrade various polysaccharides, especially plant‐derived ones such as fructans, as well as structural carbohydrates like xylan and pectin (Supporting Information Table A6). We also found evidence of pathways linked to B‐complex vitamin biosynthesis and IPP formation—an important intermediate in terpenoid biosynthesis—highlighting this isolate's potential for diverse biochemical functions (Supporting Information Table A6). In Paenibacillus sp., complete pathways were identified for the degradation of polysaccharides, including inulin, levan—linked to the production of this compound observed in the strain (Mendonça et al. 2021)—xylan, cellulose, and pectin (Figure 4). Regarding vitamin biosynthesis, Paenibacillus sp. strain 210 possesses complete pathways for B1, B3, B5, B6, B7, B9, and B12, indicating its ability to synthesize these vitamins, as well as IPP (isopentenyl diphosphate), which is associated with terpenoid production. In contrast, the pathways for B2 and K2 are incomplete, suggesting potential limitations in their biosynthesis or dependence on external sources (Figure 4).
Simplified scheme of the metabolic pathways verified in Paenibacillus sp. strain 210. Representations used are available at polysaccharides (https://www.genome.jp/kegg/compound/) and plant cell structure (https://www.swissbiopics.org/). ABC, ATP‐binding cassette; ATP, adenosine triphosphate; CAZyme, carbohydrate‐active enzyme; GTP, guanosine triphosphate; IPP, isopentenyl pyrophosphate.
In Silico Structural Comparison of Enzymes Involved in Cellulose, Pectin, and Xylan Metabolism Pathways
3.4
Functional annotation of complex carbohydrate metabolism in Paenibacillus sp. strain 210, based on EC numbers and KO terms, enabled the in silico reconstruction of cellulose, pectin, and xylan degradation pathways. Enzyme structures predicted by AlphaFold 3 were compared with experimentally solved references using FATCAT, providing mechanistic insight into the strain's polysaccharide‐degrading capacity.
Key enzymes analyzed included endoglucanase B, beta‐glucosidase A, pectinesterase A, alpha‐galacturonidase, endo‐1,4‐beta‐xylanase A, beta‐xylosidase, and xylulose kinase. Endoglucanase B (GH5, E.C. 3.2.1.4; ABI1387_25700) showed high structural conservation with a Bacillus sp. cellulase protein data bank (PDB): 5E09; with 531 aligned positions and a root‐mean‐square deviation (RMSD) of 1.06 Å, supported by 90.8% of residues in favored Ramachandran regions (Figure 5 and Supporting Information Figure A9 and Table A7). Similarly, beta‐glucosidase (GH1; ABI1387_03255) aligned with P. polymyxa (1BGG), showing 448 equivalent positions and RMSD of 1.91 Å (Supporting Information Table A7).
Molecular alignment of protein relationships with cellulose and pectin degradation pathways from Paenibacillus sp. (in green) with the references structures in the PDB (in red), reference pathway for cellulose metabolism in KEGG: map00500, and pectin in KEGG: map00040. KEGG, Kyoto Encyclopedia of Genes and Genomes.
Comparable structural conservation was observed for pectin metabolism enzymes, pemA (AB1387_16255 vs. 2NSP, Dickeya dadantii) and lplD (AB1387_04930 vs. 3FEF, B. subtilis), as well as endo‐1,4‐beta‐xylanase A (GH11, E.C. 3.2.1.8; AB1387_04845 vs. 1YAW, B. subtilis), reveal high three‐dimensional similarity (Figures 5 and 6). Ramachandran analysis shows 89.4% of residues in favored regions and 10.6% in allowed regions, confirming reliable structural models (Supporting Information Figure A9 and Table A7). The xylanase structures share 184 equivalent positions with an RMSD of 0.18 Å and no significant distortions (Supporting Information Table A7).
Molecular alignment of protein relationships with xylan degradation pathways from Paenibacillus sp. (in green) with the references structures in the PDB (in red). reference pathway for xylan metabolims in KEGG: rn00040 and RAST: https://rast.nmpdr.org/seedviewer.cgi?page=Subsystems&subsystem=Xylose_utilization. KEGG, Kyoto Encyclopedia of Genes and Genomes; RAST, Rapid Annotation of Subsystems Technology.
Plant Growth Promotion Mechanisms in Paenibacillus sp. Strain 210
3.5
The plant growth–promoting (PGP) capabilities of Paenibacillus sp. strain 210 are supported by a suite of genes involved in auxin biosynthesis, phosphate solubilization, nitrogen metabolism, and nitrogen fixation. These genetic traits enable the bacterium to enhance plant nutrient acquisition, stress tolerance, and overall growth.
IAA production, a key phytohormone for root development, involves the tryptophan‐dependent pathway with genes such as aroC, aroH, aroF, trpA–D, and ipdC, enabling chorismate and tryptophan synthesis and facilitating decarboxylation reactions critical for IAA biosynthesis (Table 3).
Phosphate solubilization in Paenibacillus sp. strain 210 is potentially supported by genes encoding phosphatases (phoN, phoA), transporters (phnC/D/E and ptsS/C/A/B), and the regulator phoU (Table 4).
Nitrogen assimilation in Paenibacillus sp. strain 210 is potentially supported by genes involved in nitrate reduction (narG/H/I/J and nirB/D), nitrate/nitrite transport (narK and nirC), and ammonium uptake (amtB) (Table 5).
The nif gene cluster in Paenibacillus sp. strain 210 (Table 6) potentially enables atmospheric nitrogen fixation. Key genes, including nifH, nifD/K, and nifB/E/N, along with hesA for cofactor biosynthesis, may allow the reduction of N₂ to NH₃, supplying plants with a direct nitrogen source.
The search for this gene set was also extended to species that are phylogenomically related to strain 210 (Supporting Information Table A8).
Discussion
4
Paenibacillus sp. strain 210, isolated from crude oil–contaminated soil in Brazil, exhibits genomic traits reflecting ecological specialization and biotechnological potential. Its genome reveals taxonomic novelty, enzymatic capabilities for polysaccharide degradation, antimicrobial biosynthesis, and PGP mechanisms, providing a foundation for understanding its adaptation to extreme environments and potential applied uses.
Genomic Basis for Taxonomic Distinction and Ecological Adaptation in Paenibacillus sp. Strain 210
4.1
Paenibacillus sp. strain 210 represents a novel species within the genus, as evidenced by phylogenomic analysis and ANI values below 93% compared with its closest relatives (P. kribbensis, P. peoriae, and P. brasilensis) (Da Mota et al. 2004; Xu et al. 2014; Do Carmo Dias et al. 2021; Yuan et al. 2022).
Considering that genomic approaches are robust enough for statistically supported taxonomic assignments—exemplified by ANI values below 95% as a species threshold (Chun et al. 2018)—and given the absence of distinctive sequences in universal bacterial markers such as 16S rRNA (Mendonça et al. 2021) or genus‐specific markers (Da Mota et al. 2004), our findings, integrated with phylogenomic analyses based on 308 single‐copy core genes shared with the closest related genomes, provide a plausible inference for the designation of a new taxon, as similarly demonstrated in other studies (Da Silva et al. 2022; Xue et al. 2023; Kong et al. 2025).
The 5.705 Mb circular genome, characterized by a GC content of 46.5% and devoid of plasmids, harbors four intact prophage regions and 13 BGCs linked to antimicrobial production. These features distinguish strain 210 from established Paenibacillus species (Supporting Information Tables A1–A4). While paenilan—a lantibiotic with anti‐Gram‐positive activity—is shared with P. polymyxa E681 (Jeong et al. 2019), its absence in other clade members underscores strain 210's phylogenetic novelty (S. H. Lee et al. 2013; Jeong et al. 2019; Tsai et al. 2022; Dobrzyński and Naziębło 2024). The strain 210 displayed nitrogen fixation (nif), AIA and phosphate solubilization genes on the chromosome in contrast to plasmid‐bearing P. polymyxa strains, suggesting niche specialization in hydrocarbon‐rich environments (Jeong et al. 2019; Do Carmo Dias et al. 2021; Yuan et al. 2022). Such genomic traits align with its isolation from crude oil–contaminated soil, positioning it as a candidate for bioremediation strategies aimed at restoring microbial communities in polluted ecosystems (Y. Li, Li, et al. 2022; Dewiyanti et al. 2024; T. Li et al. 2024; Polyak et al. 2024).
CAZymes and Structural Basis of Biomass Deconstruction
4.2
Paenibacillus sp. strain 210 encodes 259 CAZymes, surpassing related species like P. terrae (52 GHs) and P. polymyxa (Jeong et al. 2019). Key families include GH5 (cellulases), GH43 (xylanases/arabinofuranosidases), and PL9/PL10 (pectinases), which enable degradation of cellulose, xylan, and pectin—critical for processing plant biomass in degraded soils. GH43 enzymes, coupled with CBMs (CBM36/91), facilitate hemicellulose deconstruction, while PL9/PL10 pectinases may enhance root penetration in compacted soils (He et al. 2009; Paës et al. 2012; Dewiyanti et al. 2024).
The expanded GH diversity observed in strain 210 (55 families) compared with other members of the genus reflects potential ecological advantages, particularly in environments rich in complex polysaccharides derived from plant residues (Table 5). This genomic profile is consistent with the well‐documented ecological versatility of Paenibacillus spp. (Grady et al. 2016; Jeong et al. 2019; Do Carmo Dias et al. 2021; Yuan et al. 2022), which frequently inhabit the rhizosphere and contribute to organic matter turnover. The presence of PL9 and PL10 in strain 210 further underscores a distinctive enzymatic trait that may be associated with a specialized ecological niche or enhanced plant–microbe interactions, given their rare occurrence in other members of the genus (Table 5).
Although this study is based on in silico genomic analysis, the CAZyme repertoire of strain 210 provides a strategic foundation for future wet‐lab validation. Targeted enzymatic assays focusing on GH5, GH11, and PL9/10 families could assess substrate specificity, activity across environmental conditions, and synergistic lignocellulose degradation. Heterologous expression and biochemical characterization may further identify enzymes with industrial potential for biofuel production, bioremediation, and agricultural applications.
Structural modeling of the GH5 cellulase (ABI1387_25700) and GH11 xylanase (AB1387_04845) using AlphaFold 3 confirmed conserved catalytic domains (RMSD: 1.06 and 0.18 Å, respectively), validating their functional parity with homologs in Bacillus spp. (Paës et al. 2012). These enzymatic capabilities mirror P. polymyxa E681's role in root exudate processing (Jeong et al. 2019) but underscore strain 210's superior potential for lignocellulosic biorefinery applications, such as bioethanol production from sugarcane waste (He et al. 2009).
The use of structural modeling and comparison, in combination with primary functional annotation and domain‐based validation, provides a multi‐layered in silico approach to assess enzyme functionality. By integrating these steps, we enhance the reliability of our predictions regarding the potential phenotypic roles of the candidate enzymes, while acknowledging that environmental and regulatory factors also influence gene expression and phenotype, offering a robust framework for interpreting their biological significance within the context of the genome.
Gene‐Encoded Metabolic Versatility Supporting Microbial Interactions and Ecological Resilience
4.3
The genome contains complete pathways for B‐complex vitamins (B1, B3, B5, B6, B7, B9, and B12) and IPP, a terpenoid precursor. Vitamin B12 synthesis, rare in Paenibacillus, may stabilize microbial consortia in oil‐polluted soils by supporting auxotrophic organisms (Yuan et al. 2022). The strain 210 fructan metabolism (GH32 + CBM66) enables energy harvesting from plant‐derived levan, akin to P. peoriae ZBSF16 (Yuan et al. 2022), while incomplete pathways for B2 and K2 suggest metabolic dependencies that foster syntrophic interactions (Yuan et al. 2022).
It is important to note that while the genome encodes these pathways, gene presence does not necessarily imply constitutive expression. Environmental conditions, nutrient availability, interspecies interactions, and regulatory mechanisms can strongly influence whether these genes are expressed and their corresponding enzymes are functional (Bervoets and Charlier 2019; Wang et al. 2023; Sinha et al. 2025). Therefore, the metabolic versatility inferred from genomic data represents potential capabilities that require experimental validation under relevant ecological conditions. These traits, when expressed, could align with bioremediation strategies where vitamin secretion and polysaccharide degradation synergize to rejuvenate microbial communities in contaminated environments (Guo et al. 2015).
Integrative Genomic Insights Into Antimicrobial Biosynthesis and Biocontrol Functions
4.4
The BGCs in strain 210 are associated with the production of putative of antimicrobial compounds similar to fusaricidin B, paenilan, tridecaptin, and paenicidin A. Fusaricidin B, for example, is active against Fusarium oxysporum and Staphylococcus aureus (Kajimura and Kaneda 1997; Lorentz et al. 2006; S. H. Lee et al. 2013; Xu et al. 2014; Jeong et al. 2019; Kim et al. 2020; Tsai et al. 2022; Yuan et al. 2022; Dobrzyński and Naziębło 2024). It is conserved across Paenibacillus spp., while paenilan production is uniquely shared with P. polymyxa (Jeong et al. 2019). Paenicidin A, targeting L. monocytogenes (Lorentz et al. 2006), and tridecaptin, effective against multidrug‐resistant Gram‐negative pathogens, highlight dual roles in environmental remediation and pathogen suppression. These antimicrobials mirror the biocontrol traits of P. kribbensis strain t‐9 (Xu et al. 2014), P. peoriae ZBSF16 (Yuan et al. 2022) and P. polymyxa E681 (Jeong et al. 2019) but expand strain 210's utility in agroindustrial applications, such as protecting crops from soil‐borne pathogens (Dobrzyński and Naziębło 2024).
Beyond the identification of individual gene clusters, the distribution of these BGCs reveals broader evolutionary and ecological dynamics within the genus (Supporting Information Table A4). The conservation of fusaricidin B, paeninodin, and tridecaptin clusters across closely related species points to a core antimicrobial repertoire essential for competitive fitness in soil environments (Supporting Information Figures A4–A6). In contrast, the more limited occurrence of paenilan and paenicidin likely reflects recent acquisition events or selective retention, highlighting the adaptive flexibility of Paenibacillus spp. (Supporting Information Figures A7 and A8). Collectively, these features position strain 210 not only as a taxonomically relevant isolate but also as a valuable reservoir of bioactive compounds with significant biotechnological potential.
Potential PGP Mechanisms and Agricultural Prospects of Paenibacillus sp. Strain 210
4.5
The genomic characterization of Paenibacillus sp. strain 210 suggests the presence of a robust suite of PGP mechanisms, positioning it as a formidable candidate for sustainable agricultural practices. The ability of strain 210 to potentially synthesize IAA, solubilize phosphate, and fix atmospheric nitrogen is inferred from its genomic repertoire, underscores its potential to enhance crop productivity while reducing reliance on synthetic fertilizers. These traits, identified through comparative genomics and supported by existing literature on Paenibacillus species, highlight strain 210's putative adaptability and ecological versatility.
In strain 210, IAA production, predicted to be mediated by genes such as ipdC, trpA‐E and aroC/H/F, presents metabolic pathways similar to those observed in P. peoriae ZBSF16, where it can also promote root elongation through auxin synthesis (Yuan et al. 2022). IAA is known to be critical for stimulating root architecture, thereby improving nutrient and water uptake in plants. While multiple Paenibacillus strains exhibit IAA production, strain 210 distinguishes itself through its chromosomal localization of these genes, as opposed to plasmid‐borne systems. This genomic stability may indicate a heritable and reliable trait, although experimental validation is required to confirm its functionality under environmental conditions.
The strain's predicted capacity to solubilize phosphate via phoN, phoA, and phn/phs operons aligns with the functional repertoire of Paenibacillus kribensis CX‐7, which efficiently converts insoluble phosphate into bioavailable forms (Ai‐min et al. 2013). The presence of this operon (Table 4), including regulatory and transport components such as phoU, suggests a coordinated system for phosphate acquisition and homeostasis under nutrient‐limited conditions, supporting the strain's putative phosphate‐solubilizing potential. In nutrient‐poor or polluted soils, such as those contaminated by hydrocarbons, these mechanisms could mitigate phosphorus limitation and contribute to plant growth promotion and soil recovery.
Paenibacillus sp. strain 210 nif gene cluster, encoding nitrogenase and FeMo‐cofactor biosynthesis proteins, indicates the potential for atmospheric nitrogen fixation—a trait shared with P. brasilensis PB24 (Do Carmo Dias et al. 2021). However, strain 210 further complements this with genomic evidence of nitrate reduction (nar operon) and ammonium transport (amtB), creating a dual nitrogen acquisition strategy. This genetically inferred metabolic flexibility suggests nitrogen availability across diverse soil conditions, from nitrogen‐depleted agricultural lands to hydrocarbon‐rich environments.
The combination of IAA production, phosphate solubilization, and nitrogen metabolism within a single strain reflects the multifunctionality observed in recently described species such as Paenibacillus monticola sp. nov., isolated from extreme environments (H. P. Li, Gan, et al. 2022). Notably, strain 210's genomic repertoire appears specifically adapted to thrive in crude oil–contaminated soils, an underexplored niche in plant growth‐promoting rhizobacteria (PGPR) research. Comparative analysis of its gene content with species exhibiting experimentally validated, desirable traits further supports the isolate's biotechnological potential (Supporting Information Table A8).
This adaptability suggests that strain 210 could potentially serve as a “pioneer” microbe in degraded ecosystems, simultaneously rehabilitating soils and promoting plant growth—a hypothesis that warrants experimental confirmation before application in commercial PGPR formulations.
Conclusion
5
Paenibacillus sp. 210 emerges as a versatile biocatalyst, combining genomic insights and in silico structural analyses to reveal its multifaceted potential. Its CAZyme repertoire and vitamin synthesis pathways support applications in soil revitalization, bioenergy production, and residue valorization (A. P. De Souza et al. 2014), while antimicrobial BGCs offer avenues for pathogen control in agriculture (Lorentz et al. 2006; Dobrzyński and Naziębło 2024). PGP mechanisms—including IAA production, phosphate solubilization, and nitrogen fixation—address critical agricultural challenges such as nutrient limitation, soil degradation, and pathogen pressure, providing a genetically resilient alternative to chemical inputs (Ai‐min et al. 2013; Do Carmo Dias et al. 2021; H. P. Li, Gan, et al. 2022). Our genomic analyses provide evidence supporting the potential for these capabilities. Comparative genomics further highlights the adaptability of strain 210 across bioenergy, agriculture, and environmental remediation sectors. However, further studies are required to experimentally evaluate plant–microorganism interactions, process stability, and practical applicability. Field trials in contaminated soils and exploration of synergies with hydrocarbon‐degrading consortia (Y. Li, Li, et al. 2022) could be important steps in this process. Overcoming challenges such as enzymatic scalability and consortium stability will be essential to fully exploit its metabolic flexibility for sustainable agriculture and circular bioeconomy initiatives. Overall, this study highlights the untapped potential of extremophilic isolates to drive ecological and agricultural innovation.
Author Contributions
João Victor dos Anjos Almeida: conceptualization, methodology, investigation, analysis, writing – original draft, writing – review and editing. Carlos Miguel Nóbrega Mendonça: conceptualization, data collection, writing – review and editing. Leandro Marcio Moreira: methodology, writing – review and editing. Ricardo Pinheiro de Souza Oliveira: data collection, writing – review and editing, Alessandro de Mello Varani: conceptualization, methodology, supervision, writing – review and editing. Mauro de Medeiros Oliveira: conceptualization, methodology, conceptualization, data collection, methodology, investigation, analysis, supervision, writing – review and editing.
Ethics Statement
The authors have nothing to report.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Figure A1: Phylogenomic tree assembled using single‐copy genes shared between Paenibacillus reference genomes. In green highlight, the clade closest to Paenibacillus sp. used in comparative analyses.
Figure A2: Phylogenomic relationships within the clade containing Paenibacillus sp. strain 210, reconstructed with additional genomes. The analysis confirms its evolutionary placement.
Figure A3: Heatmap generated by ClusterANImap, showing the ANI index between genomes of Paenibacillus sp. and all reference genomes. Colors range from red (high similarity) to white (low similarity), with gray areas indicating the absence of a relationship. The dendrogram highlights phylogenetic clusters based on genomic similarity.
Figure A4: Synteny and collinearity relationships among Paenibacillus genomes, shared with Paenibacillus sp. strain 210 in regions associated with fusaricidin B biosynthesis. (paeninodin, tridecaptin, paenilan, paenicidin).
Figure A5: Synteny and collinearity relationships among Paenibacillus genomes, shared with Paenibacillus sp. strain 210 in regions associated with paeninodin biosynthesis.
Figure A6: Synteny and collinearity relationships among Paenibacillus genomes, shared with Paenibacillus sp. strain 210 in regions associated with tridecaptin biosynthesis.
Figure A7: Synteny and collinearity relationships among Paenibacillus genomes, shared with Paenibacillus sp. strain 210 in regions associated with paenilan biosynthesis.
Figure A8: Synteny and collinearity relationships among Paenibacillus genomes, shared with Paenibacillus sp. strain 210 in regions associated with paenicidin biosynthesis.
Figure A9: Ramachandran plot for the modeled enzymes involved in polysaccharide degradation pathways in strain 210, generated using SAVES 6.1. The plot illustrates the distribution of phi (ϕ) and psi (ψ) dihedral angles, highlighting the favored, allowed, and disallowed regions.
Table A1: Summary of reference genomes retrieved from the NCBI RefSeq database.
Table A2: Distribution of antibiotic resistance genes detected in genome analysis. Genomes that did not yield results for this analysis were not included in the table.
Table A3: Distribution of prophage regions detected in genome analysis. Genomes that did not yield results for this analysis were not included in the table.
Table A4: Results for regions linked to secondary metabolite production predicted by antiSMASH and BAGEL4.
Table A5: Distribution of CAZy terms detected in genome analysis.
Table A6: Metabolic pathway genes labeled and reconstructed by genomic profiling of strain 210.
Table A7: Labeled proteins and reference proteins used for comparisons in their primary and tertiary structure, by the predicted models.
Table A8: Distribution of genes related to plant growth promotion mechanisms in strain 210 and in the genomes of the closest species according to our phylogenomic results.
supmat.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abramson, J. , J. Adler , J. Dunger , et al. 2024. “Accurate Structure Prediction of Biomolecular Interactions With Alphafold 3.” Nature 630, no. 8016: 493–500.38718835 10.1038/s 41586-024-07487-w PMC 11168924 · doi ↗ · pubmed ↗
- 2Ai‐min, Z. , Z. Gang yong , G. Tong guo , et al. 2013. “Solubilization of Insoluble Potassium and Phosphate by Paenibacillus kribensis cx‐7: A Soil Microorganism With Biological Control Potential.” African Journal of Microbiology Research 7, no. 1: 41–47.
- 3Alcock, B. P. , W. Huynh , R. Chalil , et al. 2023. “CARD 2023: Expanded Curation, Support for Machine Learning, and Resistome Prediction at the Comprehensive Antibiotic Resistance Database.” Nucleic Acids Research 51, no. D 1: D 690–D 699.36263822 10.1093/nar/gkac 920PMC 9825576 · doi ↗ · pubmed ↗
- 4Andreopoulos, W. B. , A. M. Geller , M. Lucke , et al. 2022. “Deeplasmid: Deep Learning Accurately Separates Plasmids From Bacterial Chromosomes.” Nucleic Acids Research 50, no. 3: e 17.34871418 10.1093/nar/gkab 1115 PMC 8860608 · doi ↗ · pubmed ↗
- 5Babalola, O. O. 2010. “Beneficial Bacteria of Agricultural Importance.” Biotechnology Letters 32: 1559–1570.20635120 10.1007/s 10529-010-0347-0 · doi ↗ · pubmed ↗
- 6Bervoets, I. , and D. Charlier . 2019. “Diversity, Versatility and Complexity of Bacterial Gene Regulation Mechanisms: Opportunities and Drawbacks for Applications in Synthetic Biology.” FEMS Microbiology Reviews 43, no. 3: 304–339.30721976 10.1093/femsre/fuz 001PMC 6524683 · doi ↗ · pubmed ↗
- 7Blin, K. , S. Shaw , H. E. Augustijn , et al. 2023. “anti SMASH 7.0: New and Improved Predictions for Detection, Regulation, Chemical Structures and Visualisation.” Nucleic Acids Research 51, no. W 1: W 46–W 50.37140036 10.1093/nar/gkad 344PMC 10320115 · doi ↗ · pubmed ↗
- 8Bortolaia, V. , R. S. Kaas , E. Ruppe , et al. 2020. “Res Finder 4.0 for Predictions of Phenotypes From Genotypes.” Journal of Antimicrobial Chemotherapy 75, no. 12: 3491–3500.32780112 10.1093/jac/dkaa 345PMC 7662176 · doi ↗ · pubmed ↗