Extending Thioflavin T Fluorescence Probe to 2‑Ethenyl-benzothiazole Derivatives: Drug-like Quadruplex Ligands with Potent Antitrypanosomatid Activity
Raquel C. R. Gonçalves, Pablo Peñalver, Nina M. Allen, Efres Belmonte-Reche, Belén García-Pérez, Susana P. G. Costa, Y. Jennifer Jiang, José María Pérez-Victoria, M. Carmen Galan, M. Manuela M. Raposo, Juan Carlos Morales

TL;DR
This study develops new drug-like compounds that bind to G-quadruplex structures and show strong antiparasitic activity against Leishmania and Trypanosoma.
Contribution
The paper introduces 2-ethenyl-benzothiazole derivatives as potent and selective antitrypanosomatid agents with improved G4 binding.
Findings
Several 2-ethenyl benzothiazole derivatives showed submicromolar activity against Leishmania and Trypanosoma parasites.
Compound 2b exhibited exceptional potency and selectivity, outperforming existing antiparasitic drugs.
Biophysical studies confirmed that the derivatives strongly stabilize G4 structures, surpassing Thioflavin T.
Abstract
Thioflavin T (ThT) is a well-established fluorescence probe with selectivity for G-quadruplex (G4) structures. Over the past few years, G4 ligands have emerged as promising candidates for the development of antiparasitic agents. Building on this concept, we explored extending ThT’s benzothiazole scaffold by introducing various 2-ethenyl aromatic and heteroaromatic moieties, aiming to enhance G4 binding affinity and potential therapeutic effect. A series of benzothiazolium derivatives were synthesized and evaluated for their antiproliferative and antiparasitic activity. Several 2-ethenyl benzothiazole derivatives showed submicromolar activity against Leishmania spp. and Trypanosoma brucei parasites, with up to 200-fold selectivity over MRC-5 human lung fibroblasts. Notably, compound 2b demonstrated remarkable potency, with an IC50 of 0.48 nM and a selectivity index of 46,151 against…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1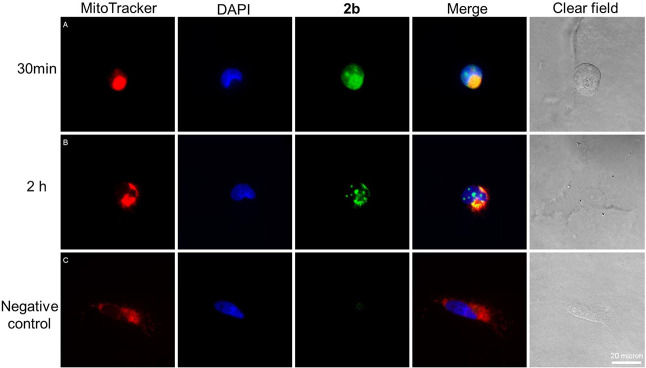 2
2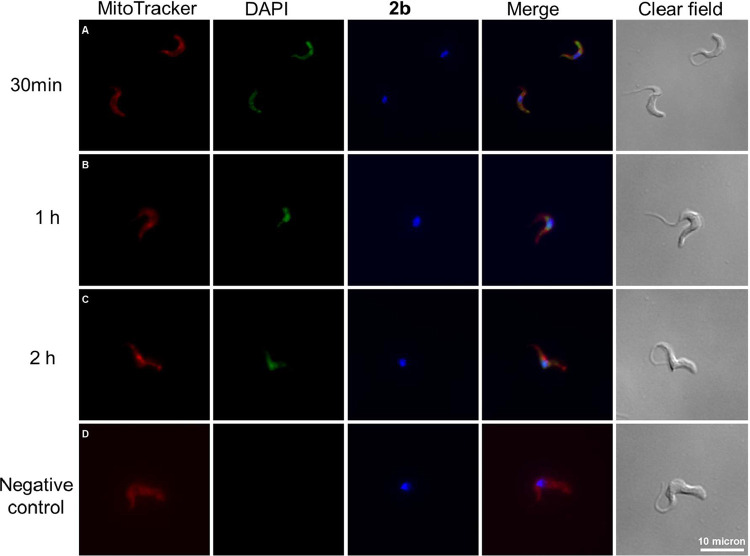 3
3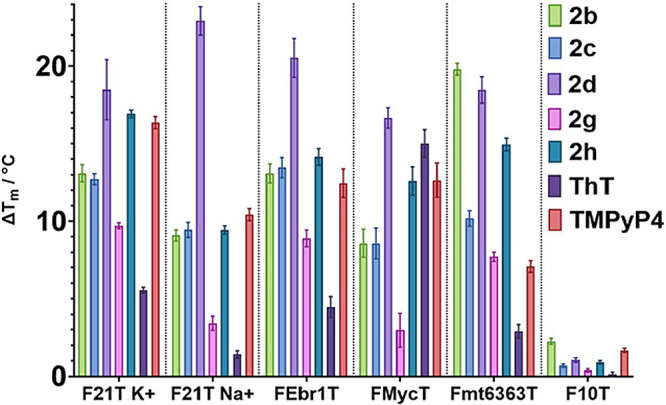 4
4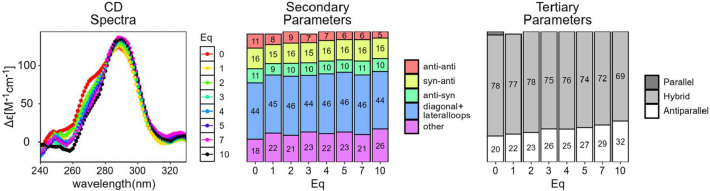 5
5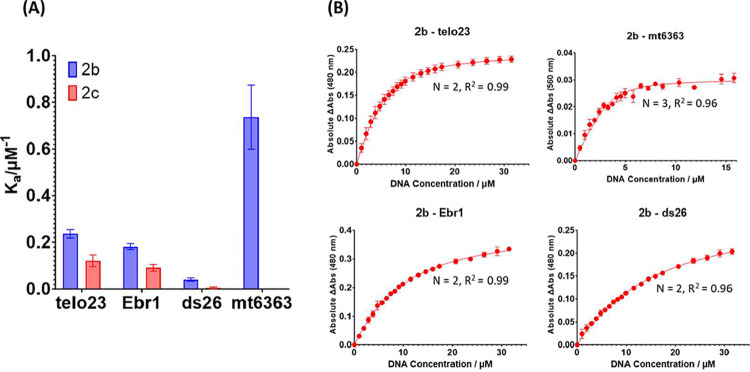 6
6| IC50 (μM) | selectivity index (SI) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| compound |
|
| MRC-5 | HeLa | HT-29 |
|
| HeLa | HT-29 |
|
| 0.37 ± 0.18 | 0.48 ± 0.11 | 2.02 ± 0.12 | 5.5 | 4.2 | ||||
|
|
|
| 1.52 ± 0.37 | 9.28 ± 0.77 | 6.56 ± 0.49 |
| 62.7 | 0.16 | 0.23 |
|
| 0.34 ± 0.13 |
| 5.13 ± 0.76 | 8.89 ± 1.59 | 8.91 ± 0.88 | 15.1 |
| 0.58 | 0.58 |
|
|
| 0.33 ± 0.017 | 3.24 ± 0.14 | 5.28 ± 0.11 | 3.79 ± 0.13 |
| 9.8 | 0.62 | 0.86 |
|
| 2.29 ± 0.31 | 0.24 ± 0.029 | 9.40 ± 1.06 | 78.01 ± 8.26 | 76.42 ± 1.44 | 4.1 | 39.6 | 0.12 | 0.12 |
|
| 0.61 ± 0.045 | 0.61 ± 0.092 | 3.68 ± 0.17 | 6.0 | 6.0 | ||||
|
|
| 0.17 ± 0.04 | 4.01 ± 0.09 | 29.08 ± 1.05 | 89.49 ± 9.55 |
| 23.9 | 0.14 | 0.04 |
|
|
|
| 0.08 ± 0.03 | 0.84 ± 0.01 | 1.00 ± 0.16 |
| 12.0 | 0.09 | 0.08 |
|
| 1.83 ± 0.06 | 0.045 ± 0.012 | 3.88 ± 0.17 | 2.12 | 86.2 | ||||
|
| 0.37 ± 0.04 | 0.99 ± 0.11 | 4.00 ± 0.14 | 10.81 | 4.0 | ||||
|
|
| 0.023 ± 0.0002 | 0.183 ± 0.038 | 38.9 | 7.9 | ||||
|
| >10 | 20.82 ± 4.86 | >25 | >1.7 | |||||
|
| 0.35 ± 0.06 | 0.044 ± 0.015 | 4.12 ± 0.52 | 11.77 | 93.64 | ||||
|
| 0.0042 ± 0.0003 | ||||||||
|
| 0.88 ± 0.02 | ||||||||
|
| 5.71 ± 0.46 | ||||||||
|
| 1.92 ± 0.05 | ||||||||
| IC50 (μM) | selectivity index (SI) | ||
|---|---|---|---|
| compounds |
| MRC-5 |
|
|
| 0.00055 ± 0.00023 | 1.52 ± 0.37 | 2,763.6 |
|
| 0.062 ± 0.003 | 0.183 ± 0.038 | 2.95 |
| IC50 (μM) |
| ||||
|---|---|---|---|---|---|
| compound |
| THP-1 | MRC-5 | THP-1 | MRC-5 |
|
|
|
| 1.52 ± 0.37 |
|
|
|
| 0.0281 ± 0.00473 | 0.93 ± 0.02 | 5.13 ± 0.76 | 33 | 182 |
|
| 0.03 ± 0.02 | 213.93 ± 3.36 | 4.12 ± 0.52 | 7,131 | 137.33 |
|
| 5.1 ± 0.56 | 55.19 ± 0.11 | 10.82 | ||
|
| 4.41 ± 1.96 | 9.89 ± 1.98 | 2.24 | ||
| properties | compound 2b | TMPyP4 | quarfloxin | |
|---|---|---|---|---|
| physicochemical | MW | 422.03 | 1362.37 | 604.26 |
| log | 4.625 | 4.448 | 5.034 | |
| TPSA (Å2) | 7.12 | 301.68 | 92.59 | |
| nHA | 2 | 20 | 9 | |
| nHD | 0 | 2 | 1 | |
| nRot | 3 | 8 | 7 | |
| medicinal chemistry | Lipinski | accepted | rejected | rejected |
| SAscore | 2.622 | 6.775 | 4.003 | |
| absorption | Caco-2 permeability (logcm/s) | –4.616 (yes) | –6.024 (no) | –5.068 (yes) |
| MDCK permeability(cm/s) | 1.9 × 10–5 (yes) | 1.2 × 10–5 (yes) | 1.6 × 10–5 (yes) | |
| distribution | VD(L/kg) | 3.16 | –0.232 | 2.44 |
| BBB penetration* | 0.511 (medium) | 1 (high) | 0.047 (low) | |
| metabolism | CYP1A2 inhibitor/substrate* | 0.946/0.936 | 0.102/0.943 | 0.149/0.79 |
| CYP2C19 inhibitor/substrate* | 0.729/0.837 | 0.051/0.057 | 0.546/0.752 | |
| CYP2C9 inhibitor/substrate* | 0.199/0.669 | 0.024/0.007 | 0.612/0.545 | |
| CYP2D6 inhibitor/substrate* | 0.864/0.883 | 0.009/0.84 | 0.888/0.876 | |
| CYP3A4 inhibitor/substrate* | 0.602/0.384 | 0.004/0.249 | 0.927/0.92 | |
| excretion | CL(mL/min/kg) | 6.93 (medium) | 0.831 (low) | 2.026 (low) |
|
| 0.215 | 0.004 | 0.053 | |
| toxicity | hERG blockers* | 0.056 (low) | 0.941 (high) | 0.963 (high) |
| H-HT* | 0.021 (low) | 0.988 (high) | 0.96 (high) | |
| AMES toxicity* | 0.983 (high) | 0.048 (low) | 0.813 (high) | |
| rat acute oral toxicity* | 0.029 (low) | 0.179 (low) | 0.354 (medium) |
- —H2020 European Research Council10.13039/100010663
- —Ministerio de Ciencia, Innovaci?n y Universidades10.13039/100014440
- —Ministerio de Ciencia, Innovaci?n y Universidades10.13039/100014440
- —Ministerio de Ciencia, Innovaci?n y Universidades10.13039/100014440
- —Global Challenges Research Fund10.13039/100016270
- —Funda??o para a Ci?ncia e a Tecnologia10.13039/501100001871
- —Funda??o para a Ci?ncia e a Tecnologia10.13039/501100001871
- —Funda??o para a Ci?ncia e a Tecnologia10.13039/501100001871
- —EPSRC Centre for Doctoral Training in Technology Enhanced Chemical Synthesis10.13039/501100018959
- —EPSRC Centre for Doctoral Training in Technology Enhanced Chemical Synthesis10.13039/501100018959
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsDNA and Nucleic Acid Chemistry · Trypanosoma species research and implications · Research on Leishmaniasis Studies
G-Quadruplexes (G4), unique secondary structures formed by guanine-rich DNA and RNA sequences, have attracted considerable attention due to evidence of their implication in many cellular and genetic processes. They are assembled by stacked planar arrangements of four guanine bases (G-tetrads) that are held together by Hoogsteen hydrogen bonds. The stability of these structures is particularly dependent on the interaction with monovalent cations, especially Na^+^ and K^+^, and small molecules, known as G4 ligands. ?,? Computational methods have been developed to predict putative quadruplex sequences (PQS) in genomic sequences. These methods employ models to analyze the sequence properties and structural features associated with G4 formation.? Through biophysical techniques such as nuclear magnetic resonance (NMR), X-ray crystallography, circular dichroism spectroscopy, Förster resonance energy transfer (FRET), and UV melting, the G4-forming capacity of specific sequences can be validated. Furthermore, chromatin-immunoprecipitation sequencing (ChIP-seq)-based mapping methods have been described for the identification and characterization of G4 structures at a genome-wide level, thus providing evidence for the existence of G4 in vitro*.*
Putative G4 motifs have been identified across the genomes of many species, including mammals, bacteria, viruses, parasite, and so forth.? Interestingly, specific genomic regions, such as promoters, enhancers, telomeres, and untranslated regions (UTRs) of mRNA, are particularly rich in G4-forming sequences. ?,?,? In fact, G4 structures have been proven to display functional roles as regulatory elements in gene expression, in DNA replication, in telomere maintenance, and in genome stability. ?,?−? ? ? G4 motifs have been explored as targets in cancer therapy due to their prevalence in tumor-related gene promoters, such as KRAS, c-MYC, c-KIT, and BCL-2,? and small molecules that selectively bind and stabilize these structures have shown potential as anticancer drugs.? In a similar way, the identification of G4 structures in the genome of pathogenic protozoa has encouraged the development of G4 ligands with promising antiparasitic activity. ?,?
Leishmaniasis, caused by Leishmania spp., and human African trypanosomiasis (or sleeping sickness), caused by Trypanosoma brucei (gambiense and rhodesiense subspecies), are neglected tropical diseases (NTDs) that have been recently proposed in the NTD roadmap for 2021–2030 by the World Health Organization, with the goal of enhancing efforts to prevent and manage these protozoan infections.? The treatment of these neglected tropical diseases is often challenging due to the significant limitations of existing antiprotozoal drugs including severe side effects, the emergence of drug resistance, and high costs. Thus, it is of great importance to develop effective antiparasitic agents. Recently, the role of G4s as transcriptional regulators in trypanosomatid parasites, namely, Trypanosoma and Leishmania spp., has been reviewed.? Studies revealed that G4 motifs could have significant regulatory functions in these protozoan pathogens, particularly as modulators of kinetoplastid DNA replication, as transcriptional regulators of epigenetic modifications, and in antigenic variation. Hence, these findings further support the idea that G4-targeting molecules may be a promising therapeutic strategy. In the last years, compounds based on naphthalene diimide, perylene diimide, stiff-stilbene, phenanthroline, quinazoline, diquinolinyl-pyridine, azobenzene, and dithienylethene cores have been reported as G4 binders that display potent antiparasitic activity. ?−? ? ? ? ? ? ? ?
Thioflavin T (ThT), a benzothiazole derivative (Figure), was first reported as a fluorescent probe for the detection of amyloid fibrils.? Decades later, Mohanty et al. demonstrated the dual role of ThT as a quadruplex folding-inducer in the 22AG human telomeric DNA and as a specific G4-fluorescence probe through an emission enhancement upon interaction with quadruplex motifs.? Moreover, ThT exhibited distinct recognition of RNA G-quadruplex structures in contrast to other forms of RNA, such as single-stranded RNA (ss-RNA) and hairpin RNA.? Other benzothiazole-based probes have been studied for the fluorescence detection of G-quadruplex structures. ?−? ?
The benzothiazole core is an important scaffold with a wide range of pharmacological properties including anticancer, anti-inflammatory, antidiabetic, anticonvulsant, antioxidant, antiviral, and antimicrobial activity.? The antiparasitic activity of derivatives based on this scaffold has also been described. ?−? ? ? ? ? ? ? ? Push–pull benzothiazole structures, with an electron-acceptor benzothiazolium moiety functionalized with electron-donor amino-substituted benzene groups through a π-conjugated bridge, have been associated with higher antimicrobial activity. ?,? More recently, Wu et al. reported a new family of benzothiazole derivatives as potential G4 ligand-based c-MYC transcription inhibitors for cancer treatment.?
In this work, we explored extending the benzothiazole scaffold of ThT with varied 2-ethenyl aromatic and heteroaromatic groups. Our plan was to investigate how different C-2 modifications of the benzothiazolium core affect their G4 binding capability and their therapeutic potential as antiproliferative and antiparasitic agents. The derivatives were evaluated in tumor cell lines (HeLa and HT-29) and nontumor cell lines (MRC-5 and THP-1), and in the parasites Leishmania major extracellular promastigotes and intracellular amastigotes, the clinically relevant form and in bloodstream forms of Trypanosoma brucei parasites. The intracellular localization and in silico ADMET profile of the best compound were also studied. Finally, we confirmed the G4 binding ability of these compounds using a variety of biophysical techniques.
Results and Discussion
Synthesis and Characterization of 2-Ethenyl Benzothiazolium Derivatives
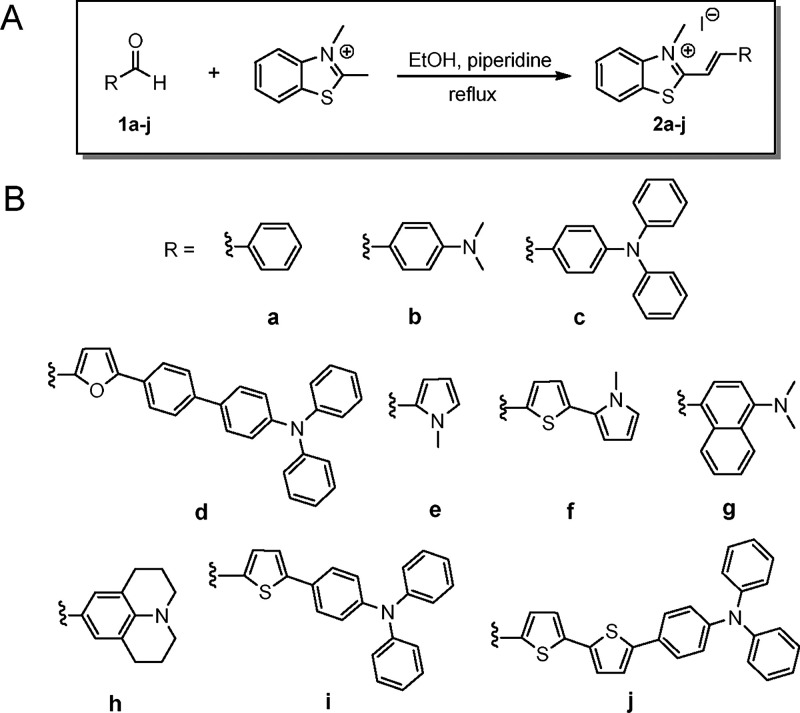
The new 2-ethenyl benzothiazolium derivatives 2d, 2f, 2i, and 2j were synthesized through the Knoevenagel condensation reaction of 2,3-dimethylbenzo[d]thiazol-3-ium iodide with the corresponding aldehydes 1d, 1f, 1i, and 1j under reflux in ethanol. All compounds precipitated from the reaction mixture and were washed with cold ethanol to give the pure benzothiazolium derivatives in moderate to low yields (Table S1). The yields obtained were low (8–30%), most probably due to the reduced reactivity of the corresponding aldehydes, which likely hindered the progress of the Knoevenagel condensation, and the difficulty in isolating the final products. In both cases, unreacted starting materials were the predominant components, suggesting incomplete conversion rather than the formation of major side products.
Experimental details and structure characterization data are described in the Supporting Information. The synthesis of benzothiazolium derivatives 2a, 2b, 2c, 2e, 2g, and 2h has been described previously by our research group and others. ?−? ? The synthesis and photophysical characterization are presented in Scheme and Table S1.
(A) Synthesis of 2-Ethenyl Benzothiazolium Derivatives 2a–j; (B) Structures of the Compounds Prepared
Antiparasitic and Anticancer Activity
To investigate the biological activity of the benzothiazolium compounds 2a–j, their toxicity was assessed in the bloodstream form of Trypanosoma brucei and promastigote and amastigote forms of L. major parasites. The antiparasitic activity of ThT, TMPyP4, and RHPS4 (G-quadruplex ligand references), suramin and fexinidazole (T. brucei positive drug controls), and miltefosine and amphotericin B (L. major positive drug controls) was also included. The antiproliferative activity was investigated in cervical carcinoma (HeLa) and colorectal adenocarcinoma (HT-29) human cancer cell lines. Cytotoxicity was evaluated against control human lung fibroblast (MRC-5) and human leukemia monocytic (THP-1) cell lines. Cell viability results were expressed as the half-maximal inhibitory concentration (IC_50_) calculated through dose–response curves, and the selectivity index (SI) was determined by the ratio of IC_50_ in healthy cells to IC_50_ in parasites or cancer cells, in order to predict the potential therapeutic window of each compound (Tables–?).
1: Antiparasitic Activity against Bloodstream Form of T. brucei and Promastigotes Form of L. major and Cytotoxicity in MRC-5, HeLa and HT-29 Represented as IC50 Values (μM) with Error Presented as σ
All tested compounds exhibited potent antitrypanosomal activity, with IC_50_ ranging five-orders of magnitude (from 0.000019 to 2.39 μM). Compounds 2b, 2d, 2g, and 2h were notably active as well as selective against T. brucei, with IC_50_ in the nanomolar range and SI above 200-fold. These compounds were considerably more active than the reference drug fexinidazole (IC_50_ = 0.88 μM). TMPyP4 and RHPS4, well-established G4 ligands known for their telomerase inhibitory activity in cancer cells, ?,? exhibited IC_50_ values >10 and 0.35 μM, respectively, both higher than those observed for the most active benzothiazolium derivatives. Importantly, the derivative functionalized with an (N,N-dimethylamino)phenyl moiety (2b) was particularly active, displaying the lowest IC_50_ (0.019 nM) together with a remarkable 79,206-fold selectivity toward the parasites. The toxicity and therapeutic window of this compound are substantially higher than other recently reported antitrypanosomal agents, ?,? as well as currently approved clinical drugs. We were also interested in investigating the antiparasitic activity of ThT, a selective G4-fluorescence probe, due to its structural similarity to our derivative 2b. We observed that the absence of the ethenyl bridge and the C-6 substitution of the benzothiazole core drastically decreased the toxicity for T. brucei compared with compound 2b (IC_50_ = 4.7 nM vs 0.019 nM, respectively). Moreover, ThT displayed higher toxicity on MRC-5 cells, resulting in lower selectivity compared with that of compound 2b.
When we compared compound 2c with 2d, 2i, and 2j, we observed that introducing a linker between the ethenyl-benzothiazolium scaffold and the triphenylamine group affected the activity and selectivity. Introducing a 5-phenylfuran linker (compound 2d) considerably increased activity and selectivity, while replacing it with a thiophene (compound 2i) or 2,2′-bithiophene (compound 2j) moiety did not show improvements.
Given the exceptionally high antitrypanosomal activity of 2b (IC_50_ in the low nM range), the CellTiter-Glo luminescent cell viability assay was used to further corroborate our results (Table). The IC_50_ values obtained with this assay (0.00055 μM) were consistent with those obtained through the resazurin methodology. Although the IC_50_ was 27-fold higher when measured by the CellTiter-Glo luminescent method, this difference is not considered significant within the variability typically associated with distinct biological assays. Overall, compound 2b demonstrated significantly greater activity and selectivity compared to those of our reference and the parent compound, ThT.
2: Antiparasitic Activity against T. brucei Was Measured by the CellTiter-Glo® Assay
Concerning antileishmanial activity, the tested compounds displayed significantly higher potency (IC_50_ ranging from 0.0065 to 0.99 μM) than the reference drug miltefosine (IC_50_ = 5.71 μM) and amphotericin B (IC_50_ = 1.92 μM), as well as than the G4 ligand TMPyP4 (IC_50_ = 20.82 μM). While RHPS4 (IC_50_ = 0.044 μM) showed higher potency than some of the tested compounds, the majority of the benzothiazolium derivatives displayed comparable or superior activity. Compound 2h was the most toxic against the parasites, with an IC_50_ of 0.0065 μM; however, the derivative 2c, substituted with a triphenylamino group, exhibited lower toxicity toward MRC-5 cell lines, resulting in a selectivity index 21-fold higher than compound 2h (SI = 252 vs 12, respectively). Furthermore, introducing different linkers between the ethenyl-benzothiazolium unit and the triphenylamine moieties generally reduced selectivity and, in most cases, also decreased toxicity. For instance, compound 2d featuring a 5-phenyl-furan group, and compound 2j, featuring a 2,2′-bithiophene group, both exhibited reduced toxicity and selectivity compared to compound 2c. Conversely, the introduction of a thiophene heterocycle (compound 2i) resulted in increased toxicity while still exhibiting selectivity lower than that of compound 2c. Moreover, comparing the antileishmanial activity of ThT with the synthesized compounds, it was observed that although it exhibited similar activity as compound 2c, the selectivity toward MRC-5 cells was considerably lower. Overall, compound 2b demonstrated the greatest therapeutic potential for bloodstream forms of T. brucei, while compound 2c displayed the best potential for promastigotes forms of L. major.
Further experiments were conducted to evaluate the activity of compounds 2b and 2c against the intracellular amastigote form of L. major (Table). Interestingly, compound 2b demonstrated significantly greater potency against the amastigotes compared to the promastigote form (IC_50_ = 0.000484 μM vs 0.02 μM, respectively). Moreover, it exhibited a remarkable selectivity index over THP-1 macrophages (SI = 46,151) and a 50-fold increase in selectivity toward MRC-5 cells when tested against the intracellular form of the parasite. In contrast, the activity of compound 2c was similar against both promastigote and amastigote forms of the parasite; however, its selectivity index was lower for THP-1 macrophages compared to MRC-5 cells (SI = 33 vs 182, respectively). Moreover, compound 2b demonstrated a 62-fold increase in activity compared to the G4 ligand RHPS4 and approximately 10,000-fold greater potency than the clinically used drugs miltefosine and amphotericin B, along with a substantially higher selectivity index.
3: Antileishmanial Activity against Amastigotes Form of L. major
The anticancer activity of the derivatives 2b, 2c, 2d, 2e, 2g, and 2h was evaluated. However, all the compounds demonstrated lower activity compared to the activity against the parasites and poor selectivity for the cancer cells over the noncancerous MRC-5 cell line (SI < 1).
Subcellular Localization of Derivative 2b
The uptake and subcellular localization of 2b in MRC-5 cells and T. brucei parasites were investigated through fluorescence microscopy, taking advantage of the intrinsic fluorescence of 2b. After 30 min of incubation, the compound was internalized into both types of cells, exhibiting a nonspecific intracellular distribution, with fluorescence detected in both the cytoplasm and mitochondria (FiguresA and ?A). At longer periods of incubation, the fluorescence of compound 2b showed increased colocalization in the nucleus and kinetoplast, suggesting that the compound specifically localizes in these organelles over time (FiguresB and ?B,C). These findings are particularly significant, as DNA G-quadruplexes mainly form in the nucleus and kinetoplast, and mitochondria, emphasizing the importance of the ability of 2b to reach and accumulate within these specific organelles.
Fluorescence microscopy of MRC-5 cells treated with compound 2b. Cells were incubated with 5 μM compound 2b (green channel) for 30 min (A) and 2 h (B). Mitochondria staining was performed with MitoTracker deep red (red channel), and nuclear staining was performed using green nuclear dye (blue fluorescence). Negative control is displayed in row (C). Scale bar: 20 μm.
Fluorescence microscopy of T. brucei parasites treated with compound 2b. Parasites were incubated with 5 μM compound 2b (green channel) for 30 min (A); 1 h (B); and 2 h (C). Mitochondria staining was performed with MitoTracker deep red (red channel) and nuclear staining was performed using Green nuclear dye (blue fluorescence). Negative control is displayed in row (D). Scale bar: 10 μm.
In Silico ADMET Properties
Given its strong therapeutic activity and in line with our focus on the benzothiazole scaffold as a pharmacophore to improve the drug-like nature of G4 ligands, we calculated the ADMET (absorption, distribution, metabolism, excretion/toxicity) profile of compound 2b and compared the results with other commercial G4 ligands, including the porphyrin TMPyP4 and quarfloxin, a fluoroquinolone-based G4 ligand that progressed to Phase II clinical trials for neuroendocrine tumors (ClinicalTrials.gov identifier: NCT00780663),? using the ADMET lab2.0 online server? (Table).
4: ADMET Properties of Compound 2b and G4 Ligands TMPyP4 and Quarfloxin
The analysis revealed compound 2b’s Lipinski’s rule compliance, suggesting favorable oral bioavailability as well as a low synthetic accessibility score. In contrast, TMPyP4 and quarfloxin violated Lipinski’s criteria and presented greater synthetic complexity. Compound 2b and quarfloxin exhibited adequate permeability and optimal blood distribution, whereas TMPyP4 showed reduced oral absorption and distribution profiles. The BBB permeability predictions, particularly relevant for treating stage 2 of the sleeping sickness when the central nervous system (CNS) has been compromised by the parasite, revealed compound 2b’s higher BBB penetration compared to quarfloxin, while TMPyP4 exhibited the highest BBB permeability among the three. In fact, structurally related benzothiazolium derivatives have already been reported to efficiently cross the BBB in vivo, thereby further supporting our in silico results.? Compound 2b, as well as quarfloxin, exhibited a significant likelihood of interaction with CYP450 isozymes, while TMPyP4 showed a lower overall interaction probability. Moreover, compound 2b showed higher clearance and a longer half-life than TMPyP4 and quarfloxin, indicating sustained therapeutic effects with a potentially reduced dosing frequency. Finally, toxicity assessments suggested lower hERG inhibition and hepatotoxicity for 2b than those for TMPyP4 and quarfloxin. However, the AMES indicated a higher mutagenicity risk for 2b and quarfloxin relative to TMPyP4. Acute toxicity was predicted to be low for 2b and TMPyP4 and to be high for quarfloxin.
Overall, compound 2b exhibits favorable ADMET calculated properties, particularly in terms of oral bioavailability and systemic distribution, including adequate BBB penetration, supporting its potential as a candidate for further development. To strengthen the reliability of the ADMET predictions, additional computational tools were employed alongside the primary software, namely, pkCSM and SwissADME (Table S2–S3). The comparative analysis revealed a high degree of consistency among the results, thereby supporting the reliability of the in silico predicted properties.
Binding to DNA G4
After evaluating the therapeutic activity of the 2-ethynyl benzothiazolium derivatives and identifying compounds 2b, 2c, 2d, 2g, and 2h as promising antiparasitic agents, we investigated whether the in vitro antiparasitic activity of these derivatives could be associated with their G4 binding capability. This interest was driven by their structural similarity to ThT, a well-established G4-fluorescence probe. TMPyP4 was used as a reference G4 ligand.
The stabilization of G4 structures was initially evaluated by using the Förster resonance energy transfer (FRET) melting assay. This analysis evaluated the binding of the compounds against four G4-forming oligonucleotides and a hairpin duplex sequence to determine both G4:duplex and G4:G4 selectivity. The sequences used in this study (Table S4) included the human telomeric G4, found in T. brucei and several mammalian species,? in potassium buffer (F21T K^+^, mixed parallel/hybrid G4) and sodium buffer (F21T Na^+^, antiparallel G4), a polymorphic G4 found in T. brucei ? (FEbr1T, mixed G4 topology), the c-Myc promoter G4 (FmycT, parallel G4), a human mitochondrial G4 (Fmt6363T, hybrid G4), and a hairpin duplex DNA sequence (F10T). These G4 sequences were selected to represent a possible wide variety of G4 topologies present in the genome of T. brucei. Moreover, a duplex DNA sequence was also selected in order to study the potential selective binding of the compounds to either G4 or duplex DNA. The results are summarized in Figure and Table S4.
FRET melting assays for compounds 2b, 2c, 2d, 2g, and 2h, ThT at 10 μM and TMPyP4 (1 μM as control) with different G-quadruplexes (F21T, FEbr1T, FMycT, and Fmt6363T) and duplex sequence (F10T).
The first general trend that can be observed is that all the 2-ethynyl benzothiazolium derivatives examined bind across all the G4s screened, except for 2g, which was more selective and did not show binding to F21T Na^+^ and FMycT G4s. Moreover, all the compounds were better G4 binders than control ThT, except when binding the parallel G4 FMycT, where ThT showed similar binding to 2d and 2h. Among the 2-ethynyl benzothiazolium derivatives, compound 2d showed the highest stability in the series for all G4s. 2d turns out to be the largest and most hydrophobic among them. At the same time, all benzothiazolium analogues demonstrated selectivity for G4 structures over the duplex DNA F10T.
In more detail, for the G4 structure F21T in potassium buffer (F21T K^+^), the results indicated that most compounds exhibited significant stabilization (ΔT m > 10 °C), with compounds 2d and 2h showing the highest stabilization values (ΔT m = 18.5 and 16.9 °C, respectively). In sodium buffer (F21T Na^+^), compound 2d demonstrated the highest G4 stabilization of all of the other tested analogues. A similar trend was observed with the unique T. brucei G4 sequence (FEbr1T), where compound 2d exhibited the highest stabilization value (ΔT m = 20.5 °C), while compounds 2b, 2c, and 2h (ΔT m = 13.1, 13.5, and 14.1 °C, respectively) achieved moderate stabilization levels. The induced stabilization of G4 FMycT followed a very similar pattern. However, ThT showed better binding in this sequence compared to other G4 structures, with ΔT m = 15.0 °C vs ΔT m = 1.4–5.5 °C for the other examined G4s. Lastly, for mitochondrial G4 (Fmt6363T), compounds 2b, 2d, and 2h exhibited the highest stabilization. Notably, compound 2b showed a marked preference for stabilizing the mitochondrial G4, with a ΔT m of 19.8 °C compared to 8.5–13.1 °C for other G4 sequences. Additionally, FRET analysis was conducted for compound 2b with the mt6363 sequence under increasing concentrations of potassium buffer (Figure S1). The results indicated that the level of G4 stabilization by compound 2b increased with higher potassium concentrations.
To further investigate the DNA stabilization potential and effects on the topology of ligand 2b on G4s and duplex structures, circular dichroism (CD) spectroscopy was employed on four distinct oligonucleotide sequences: telo23, mt6363, Ebr1, and ds26 (Figures and S2, S3 and Table S6). Quantitative changes in the secondary and tertiary structures of the G4 motifs upon increasing concentrations of compound 2b were assessed using principal component analysis (PCA) and singular value decomposition (SVD) of the CD data.? The results indicated that the incremental addition of 2b induces subtle alterations in the tertiary topology of the analyzed G4 structures. Specifically, while the hybrid topology of telo23 is maintained, higher concentrations of 2b promote an increase in the proportion of antiparallel G4 structures (Figure). For the mt6363 and Ebr1 G4s, ligand binding shifts the topology toward the parallel form, diminishing the prevalence of the hybrid topology observed in the absence of the compound (Figure S2). In contrast, the effect of 2b on the secondary parameters of G4s was less pronounced.
Left, CD spectra of telo23 with different equivalents of compound 2b (vertical color scale on the side). Buffer = 100 mM potassium phosphate, pH 7.4. Oligonucleotide concentration = 5 μM; center and right, SVD analysis of the influence of the number of equivalents of 2b on the secondary and tertiary parameters.
Ultraviolet–visible (UV–vis) absorbance spectroscopy titrations were carried out to quantitatively assess the binding affinity of compounds 2b and 2c to G4 sequences (telo23, ebr1, and mt6363) and the duplex sequence (ds26) (Figures and S4). Consistent with the FRET analysis, compound 2b exhibited a higher affinity for the mt6363 G4 structure compared with the other G4 sequences. Furthermore, the analysis confirmed its selectivity for G4 structures over duplex DNA.
(A) Equilibrium binding constants calculated for compounds 2b and 2c with several G-quadruplexes (telo23, Ebr1, and mt6363) and a duplex DNA (ds26) sequence. (B) UV–vis titration of 2b with telo23, mt6363, Ebr1, and ds26. Buffer = 100 mM potassium phosphate, pH 7.4. Ligand concentration: 10 μM.
Conclusions
In this research, we explored the structural extension of the benzothiazole scaffold in the ThT probe by introducing various 2-ethenyl aromatic and heteroaromatic moieties. Our findings demonstrated that these C-2 modifications improved both the G4 binding affinity and the biological activity of the resulting benzothiazolium derivatives. Specifically, we observed that the derivative functionalized with the (N,N-dimethylamino)phenyl moiety, compound 2b, exhibited remarkable activity against bloodstream forms of T. brucei (IC_50_ = 0.019 nM), 46,000-fold more active than the reference drug fexinidazole, along with an exceptional selectivity index of 79,206. To the best of our knowledge, this represents one of the most potent antitrypanosomal agents reported, with an outstanding therapeutic window. Furthermore, a comparison with ThT highlighted the critical role of the ethenyl bridge and the absence of C-6 substitution in significantly enhancing antitrypanosomal activity. For antileishmanial activity, although the julolidine-substituted derivative 2h exhibited the highest potency against L. major promastigotes, the triphenylamine-substituted 2c demonstrated greater therapeutic potential due to its selectivity index (SI = 252) and greater antileishmanial activity than miltefosine and AmB. In the L. major amastigotes assays, compound 2b exhibited outstanding potency and selectivity, with an IC_50_ of 0.48 nM and an SI of 46,151, markedly outperforming 2c as well as the standard antileishmanial drugs miltefosine and AmB. Furthermore, the predicted ADMET properties suggest that compound 2b has a more favorable drug-like profile than the reference G4 ligands, reinforcing its potential as a promising candidate for further development.
G4 binding studies using FRET-melting, CD titrations, and UV–vis titrations showed that the most active benzothiazolium derivatives also exhibit significant G4 stabilization and selectivity over duplex DNA. Among them, compound 2d displayed the highest stabilization across all tested G4 motifs, which may be attributed to its larger size and increased hydrophobicity. In contrast, ThT showed comparatively lower stabilization of these secondary structures. Fluorescence microscopy studies on compound 2b confirmed its intracellular localization mainly within the nucleus and kinetoplast, supporting the hypothesis that G4 structures within the parasite genome could serve as potential molecular and therapeutic targets. Although the most active derivatives exhibit strong DNA-G4 binding, other factors such as membrane permeability, access to the nucleus, and access to G-quadruplexes when formed within the nucleus (and/or alternative mechanisms of action such as interactions with RNA-G4s), may also affect the compounds’ activity, toxicity, and efficacy as potential antiparasitic treatments. Overall, these results highlight the potential of benzothiazolium derivatives as G4-targeting ligands and antiparasitic agents, while emphasizing the need for further investigations into their cellular uptake and broader biological effects to fully elucidate their mechanism of action.
Methods
Antiparasitic Activity
The antitrypanosomal activity of the compounds against the bloodstream form of T. brucei was evaluated using the alamarBlue assay (ThermoFisher scientific). ?,? The stock solutions of the compounds were prepared in DMSO, with the final DMSO percentage in each well maintained below 1%. 2 × 10^4^ parasites per mL were incubated at 37 °C, 5% CO_2_ in 96-wells plates (50 μL/well) either alone or in the presence of increasing concentrations of compounds for 72 h. After the incubation period, 20 μL of resazurin solution (110 ng/mL) was added to each well, and the parasites were further incubated for 4 h at 37 °C. Subsequently, cell lysis was performed using 50 μL per well of 3% SDS. The plate was then incubated at 37 °C for an additional hour, followed by measurement of fluorescence intensity using an Infinite F200 plate reader (Tecan Austria, GmbH). The excitation wavelength was set at 550 nm, and emission was recorded at 590 nm. The results are expressed as the concentration of the compound that reduces cell growth by 50% compared to untreated control cells (IC_50_). Data were presented as the average of at least three independent measurements, each conducted under triplicate conditions.
The antileishmanial activity of the compounds against the promastigote form of L. major, the clinically relevant form was determined using an MTT-based assay (Sigma-Aldrich).? Stock solutions of the compounds were prepared in DMSO with the final DMSO percentage in each well maintained below 1%. 4 × 10^6^ parasites per mL were incubated at 28 °C in 96-well plates (50 μL/well) either alone or in the presence of increasing concentrations of compounds DMSO for 72 h. After the incubation period, 10 μL of MTT (5 mg/mL) was added to each well, and the parasites were incubated for 4 h at 28 °C. Subsequently, cell lysis was performed using 50 μL/well of 20% SDS. The plate was then incubated at 37 °C for an additional hour, and then absorbance was measured at a wavelength of 540 using an Infinite F200 plate reader (TECAN Austria, GmbH). The IC_50_ was calculated as described above. Data were presented as the average of at least three independent measurements, each conducted under triplicate conditions.
The antileishmanial activity of the compounds against the intracellular form of L. major was evaluated using the Luciferase Assay System kit from Promega. THP-1 cells were seeded at a density of 3 × 10^5^ cells/mL in 96-well plates (100 μL/well) and treated with 20 ng/mL PMA (phorbol 12-myristate 13-acetate, Sigma) for 48 h to induce differentiation. Postdifferentiation, the cells were washed, and fresh medium was added (100 μL/well) before being reincubated. Following an additional 24 h, the wells were washed, and 50 μL of L. major parasites at a density of 1.5 × 10^6^ parasites/mL (suspended in RPMI-1640 medium 5% hiFBS and 5% penicillin/streptomycin) were added to the wells. The plates were then incubated at 35 °C in 5% CO_2_. After 24 h, the wells were washed three times with PBS, and fresh medium with increasing concentrations of compounds was added (100 μL/well). After another 72 h of incubation, the cells were then treated with 25 μL of lysis buffer and stored at −80 °C. Following 24 h, 25 μL of luciferase enzyme substrate was added to each well, and the luciferase activity of the amastigotes was determined using a TECAN Infinite F200 microplate reader. Results are reported as the concentration of compounds that reduce parasite growth by 50% compared to untreated control-infected macrophages (IC_50_). Data were presented as the average of at least three independent measurements, each conducted under triplicate conditions.
Controls for the AlamarBlue, MTT, and Cell Tilter assays were run at the different compound concentrations to assess optical interference. Detectable background (fluorescence and absorbance) was limited to some compounds (mainly, 2b and 2h) at 100 μM and was negligible at ≤1 μM (all IC_50_ determinations).
Cell Titer Glo Luminescent Cell Viability Assay
The trypanocidal activity of the more active compounds was also assessed by the Cell Titer Glo assay (Promega). To do so, 50 BSF T. brucei parasites per well were incubated in 96-wells plates in the presence of increasing concentrations of compounds for 72 h at 37 °C. Ten μL portion of Cell Titer Glo was then added to every well, and after 10 min of incubation at room temperature, the luminescence of the plates was recorded using the Infinite F200 plate reader (TECAN Austria, GmbH). The results herein are expressed as the compound concentration that reduces cell growth by half versus untreated control cells (IC_50_) using SigmaPlot (Four Parameter Logistic Curve). Presented data are the mean value of three independent measurements, all of them conducted under triplicate conditions.
Cytotoxicity
The cytotoxic effect of the compounds was evaluated in MRC-5, THP-1, HeLa, and HT-29 cell lines, through the alamarBlue assay (ThermoFisher scientific). ?,? Stock solutions of the compounds were prepared in DMSO, with the final DMSO concentration in each well maintained below 1%. Prior to compound addition, 5 × 10^3^ cells per mL (MRC-5, HeLa or HT-29 cells) were seeded in 96-well plates (100 μL/well) and incubated for 24 h at 37 °C, 5% CO_2_ to allow cell attachment. Subsequently, compounds were added at various concentrations ranging from 0 to 100 μM, and the plates were incubated for 72 h. For THP-1, cells were seeded at a density of 3 × 10^5^ cells/mL in 96-well plates (100 μL/well) and treated with 20 ng/mL PMA (phorbol 12-myristate 13-acetate, Sigma) for 48 h. Postdifferentiation, the cells were washed, and fresh medium was added (100 μL/well) before being reincubated. Following another 48 h, the wells were washed again, and fresh medium with increasing concentrations of compounds was added (100 μL/well). After the incubation period of 72 h, 20 μL of resazurin solution (110 ng/mL) was added to each well, and the cells (MRC-5, THP-1, HeLa, and HT-29) were incubated for an additional 4 h at 37 °C. Subsequently, cells were lysed with 50 μL of 3% (for MRC-5, HeLa, and HT-29 cells) or 20% (for THP-1 cells) SDS, which was added to each well. The plates were incubated at 37 °C for an additional hour, and subsequently, the fluorescence intensity was measured using the Infinite F200 plate reader (TECAN Austria, GmbH), exciting at 550 nm and recording the emission at 590 nm. The obtained results were expressed as the concentration of compound that reduces cell growth by 50% compared to untreated control cells (IC_50_). Data are presented as the average of at least three independent measurements, each conducted under triplicate conditions.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Burge S.Parkinson G. N.Hazel P.Todd A. K.Neidle S.Quadruplex DNA: Sequence, Topology and Structure Nucleic Acids Res.200634195402541510.1093/nar/gkl 65517012276 PMC 1636468 · doi ↗ · pubmed ↗
- 2Varshney D.Spiegel J.Zyner K.Tannahill D.Balasubramanian S.The Regulation and Functions of DNA and RNA G-Quadruplexes Nat. Rev. Mol. Cell Biol.202021845947410.1038/s 41580-020-0236-x 32313204 PMC 7115845 · doi ↗ · pubmed ↗
- 3Puig Lombardi E.Londoño-Vallejo A.A Guide to Computational Methods for G-Quadruplex Prediction Nucleic Acids Res.202048111510.1093/nar/gkz 109731754698 PMC 6943126 · doi ↗ · pubmed ↗
- 4Mergny J.-L.Phan A.-T.Lacroix L.Following G-quartet Formation by UV-spectroscopy FEBS Lett.19984351747810.1016/S 0014-5793(98)01043-69755862 · doi ↗ · pubmed ↗
- 5De Rache A.Mergny J.-L.Assessment of Selectivity of G-Quadruplex Ligands via an Optimised FRET Melting Assay Biochimie 201511519420210.1016/j.biochi.2015.06.00226079222 · doi ↗ · pubmed ↗
- 6Hänsel-Hertsch R.Spiegel J.Marsico G.Tannahill D.Balasubramanian S.Genome-Wide Mapping of Endogenous G-Quadruplex DNA Structures by Chromatin Immunoprecipitation and High-Throughput Sequencing Nat. Protoc 201813355156410.1038/nprot.2017.15029470465 · doi ↗ · pubmed ↗
- 7Marsico G.Chambers V. S.Sahakyan A. B.Mc Cauley P.Boutell J. M.Antonio M. D.Balasubramanian S.Whole Genome Experimental Maps of DNA G-Quadruplexes in Multiple Species Nucleic Acids Res.20194783862387410.1093/nar/gkz 17930892612 PMC 6486626 · doi ↗ · pubmed ↗
- 8Qi T.Xu Y.Zhou T.Gu W.The Evolution of G-Quadruplex Structure in m RNA Untranslated Region Evol. Bioinform. Online 20211711769343211035110.1177/11769343211035140 PMC 829988434366661 · doi ↗ · pubmed ↗