Mucinous Tubular and Spindle Cell Carcinoma of the Kidney: A Rare Renal Neoplasm—Case Report and Literature Review
Ionuţ Burlacu, Mariana Aşchie, Mădălina Boşoteanu, Gabriela Izabela Bălţătescu, Alexandra Dinu

TL;DR
This paper reports a rare case of mucinous tubular and spindle cell carcinoma of the kidney, emphasizing the importance of combining imaging, histopathology, and immunohistochemistry for accurate diagnosis.
Contribution
The contribution is a detailed case report and literature review highlighting diagnostic challenges and management of a rare renal tumor.
Findings
The tumor was diagnosed as MTSCC based on clinical, radiological, histopathological, and immunohistochemical features.
The patient remained disease-free at five years following surgical excision and follow-up.
Molecular profiling is suggested to differentiate MTSCC from other renal tumors and identify aggressive variants.
Abstract
Background and Clinical Significance: Mucinous tubular and spindle cell carcinoma (MTSCC) is an uncommon subtype of renal cell carcinoma, representing 1–4% of epithelial renal tumors. It usually shows a low-grade morphology and indolent behavior, although sarcomatoid variants with an aggressive course have been described. Because of its overlap with papillary renal cell carcinoma (papRCC), sarcomatoid RCC, mesenchymal tumors, and oncocytic neoplasms, diagnosis requires the integration of imaging, histopathology, and immunohistochemistry. Case Presentation: We report a 71-year-old female who presented with a three-month history of right-sided lumbar pain and intermittent hematuria. Her laboratory tests were unremarkable. Contrast-enhanced CT revealed a well-circumscribed nodular lesion in the mid-portion of the right kidney, measuring 50 × 47 × 52 mm. The patient underwent right…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
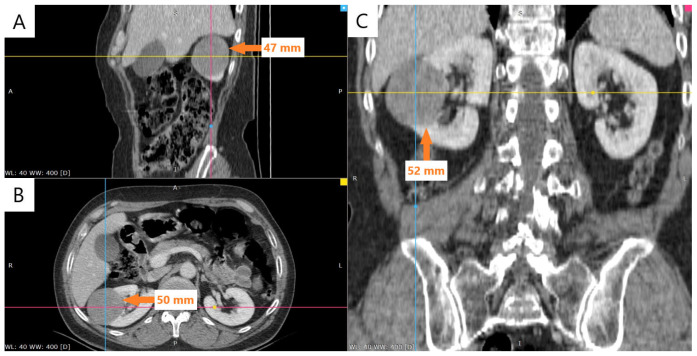 Figure 1
Figure 1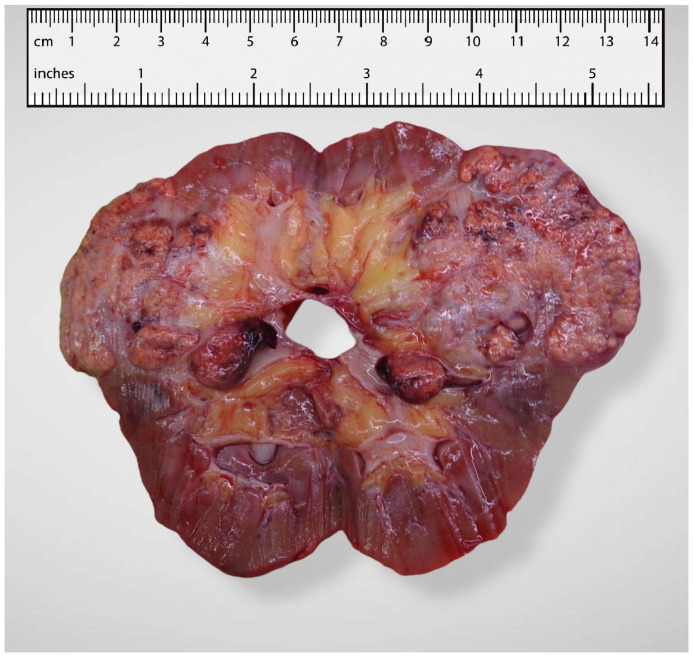 Figure 2
Figure 2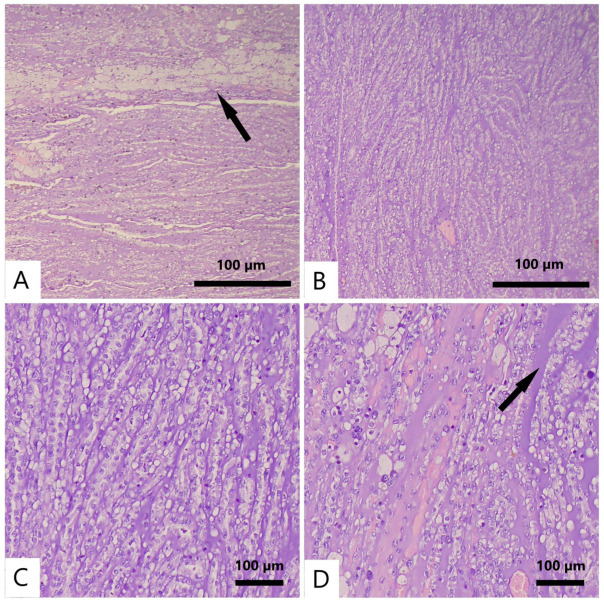 Figure 3
Figure 3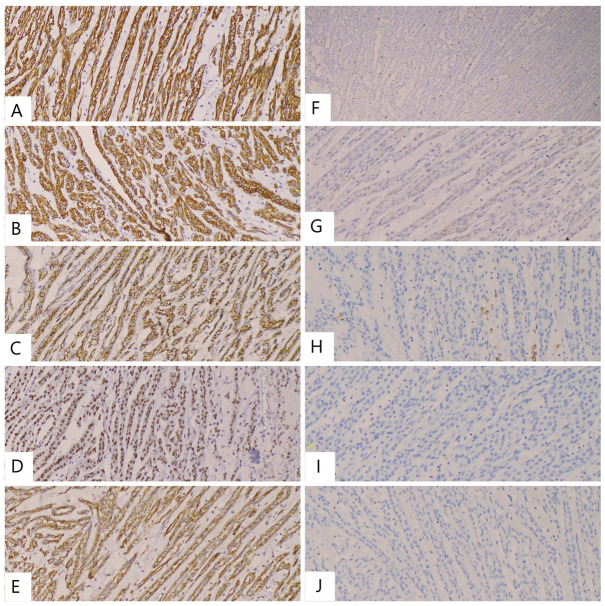 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRenal cell carcinoma treatment · Renal and related cancers · Bladder and Urothelial Cancer Treatments
1. Introduction and Clinical Significance
Mucinous tubular and spindle cell carcinoma (MTSCC) represents an uncommon form of renal cell carcinoma (RCC), initially identified as a distinct pathological entity in the 2004 World Health Organization (WHO) classification of renal tumors and subsequently maintained in later editions, including the most recent 2022 update [1,2,3].
Histologically, MTSCC is characterized by elongated tubules and spindle-shaped cells embedded within a mucinous or myxoid stroma, typically with a low nuclear grade [4]. Morphological patterns, such as mucin-poor areas, focal papillary patterns, foamy macrophages, or necrosis, may complicate diagnosis and broaden the histologic spectrum [5,6]. Before its formal classification, tumors with overlapping features were frequently misdiagnosed as unclassified RCC, papillary RCC (papRCC) with atypical features, sarcomatoid carcinoma, or metanephric adenoma [7].
Radiologically, MTSCC often shows a distinctive profile. On computed tomography (CT) and magnetic resonance imaging (MRI), it typically exhibits slow, progressive contrast enhancement and intermediate-to-high T2 signal intensity with restricted diffusion. These features differ from the rapid wash-in and wash-out enhancement pattern of clear cell RCC and the homogeneous low enhancement typical of papRCC [8,9,10].
Immunohistochemically, MTSCC is usually positive for CK7, CK19, EMA, and AMACR (P504S), while CD10 expression is absent or weak. By contrast, papRCC typically shows strong and diffuse CD10 positivity [11]. PAX8 confirms renal origin, and a low Ki-67 index (<10%) supports its indolent nature [12]. Genomic studies have shown that MTSCC is characterized by multiple chromosomal deletions, while lacking the typical gains of chromosomes 7 and 17 that are commonly observed in papRCC [13,14,15,16].
Clinically, MTSCC is generally indolent and associated with favorable outcomes after surgical excision, although high-grade or sarcomatoid variants have been reported with a poor prognosis [2,5].
In this report, we describe a case of MTSCC located in the middle third of the right kidney, emphasizing clinical, imaging, histopathological, and immunohistochemical findings, together with molecular correlations, within the context of the contemporary literature.
2. Case Presentation
2.1. Clinical Presentation
A 71-year-old female patient with a known history of chronic obstructive pulmonary disease (COPD) and bronchial asthma was referred to our department with a three-month history of right-sided lumbar pain and intermittent hematuria. Due to persistent symptoms, the patient was admitted to the Urology Clinic of the “Saint Apostle Andrew” County Emergency Clinical Hospital of Constanţa for further diagnostic evaluation and specialized treatment.
The laboratory profile was within normal limits. Abdominal and pelvic CT, performed in non-contrast and contrast-enhanced phases, showed a right kidney measuring 90 mm in bipolar diameter with a heterogeneous nephrogram. This aspect was attributed to the presence of a well-defined, nodular lesion located in the mid-portion of the kidney, protruding beyond the renal contour. The lesion appeared isodense on the non-contrast series and demonstrated low contrast enhancement, measuring approximately 50 × 47 mm in axial dimensions and 52 mm craniocaudally (Figure 1).
Based on clinical and imaging findings, a diagnosis of a right renal tumor was established. A surgical approach was deemed necessary, and the patient subsequently underwent a right open nephrectomy.
The patient’s preoperative and postoperative laboratory results are summarized in Table 1.
2.2. Pathological Findings
The nephrectomy specimen was received for gross pathological examination and measured 11 × 8 × 4 cm. At the mid-portion of the kidney, extending toward the upper pole, a well-circumscribed nodular lesion was identified, measuring 5.2 × 5 × 4 cm. The mass was encapsulated, displayed a yellowish-gray cut surface, and did not exhibit invasion into the renal pelvis (Figure 2).
A microscopic examination of hematoxylin and eosin (H&E)-stained sections revealed an epithelial neoplasm composed of cuboidal to oval cells with scant, lightly eosinophilic or clear cytoplasm and small nuclei showing mild nuclear pleomorphism. Spindle-shaped cells lacking prominent nucleoli and atypical mitotic figures were also present. The tumor was arranged in parallel cords and elongated, occasionally compressed tubular structures embedded within a mucinous stroma. Focal necrosis and clusters of foamy macrophages were noted. There was no evidence of angiolymphatic, perineural, capsular, perirenal fat, renal sinus, renal vein, or ureteral invasion. Regional lymph nodes were not identified. Representative microscopic features are shown in Figure 3.
The definitive diagnosis was established following an immunohistochemical (IHC) analysis, the results of which are summarized in Table 2; the detailed immunohistochemistry panel is available in the Supplementary Materials (Table S1).
The tumor cells demonstrated IHC positivity for CK19, CK7, EMA, PAX8, and AMACR. The Ki-67 proliferation index was quantified at <10% (approximately 5%). Conversely, IHC markers including CD117, chromogranin, RCC, and CD10 showed negative reactivity (Figure 4). These findings confirmed the final diagnosis of mucinous tubular and spindle cell carcinoma, pT1b, with low malignant potential, consistent with the initial microscopic impression.
The differential diagnosis included several renal neoplasms. Papillary RCC was ruled out due to the presence of a mucinous stroma and the absence of a distinctive papillary architecture. Sarcomatoid RCC was excluded based on the lack of marked nuclear pleomorphism, which is a hallmark of high-grade malignancy. Mesenchymal tumors were also considered; however, these are only rarely positive for cytokeratins. Oncocytoma and chromophobe RCC were excluded by their typical positivity for CD117, which was absent in this case. Neuroendocrine neoplasms were also part of the differential, but they were excluded by the lack of immunoreactivity for conventional neuroendocrine markers such as chromogranin [17,18].
At the most recent follow-up, five years after surgery, the patient remained asymptomatic and free of recurrence or metastasis.
3. Discussion
Mucinous tubular and spindle cell carcinoma accounts for only 1–4% of epithelial renal tumors, making it an uncommon subtype of renal cell carcinoma. The disease occurs predominantly in women, with a reported female-to-male ratio of 2–4:1 and is typically diagnosed at a median age of 50 years [5,16]. While most cases exhibit indolent behavior, aggressive forms—particularly those with sarcomatoid transformation—have also been reported [15,19]. Fewer than 100 cases of MTSCC are reported in the literature to date [20,21]. In a recent institutional series of 22 patients, the authors highlighted its female predominance, low proliferative index, and indolent clinical course [9], findings consistent with our case, which involved a female patient with low Ki-67 expression and an uneventful five-year follow-up.
In our patient, the lesion was well-circumscribed and located in the middle third of the kidney. CT imaging demonstrated minimal arterial enhancement with more pronounced venous phase uptake, a pattern consistent with previously described radiologic features of MTSCC [5].
A histopathological evaluation revealed the typical features of MTSCC, with cords and tubules of cuboidal cells blending into spindle cell areas within a mucin-rich stroma, along with focal necrosis and foamy macrophages [5,22]. Our case was staged as pT1b, in line with the early-stage tumors reported in both small institutional series and larger cohorts, in which pT1 disease consistently showed an indolent course [9,23]. This parallels the favorable outcome observed in our patient. The differential diagnosis included papillary RCC (excluded by the presence of mucinous stroma and absence of papillary architecture), sarcomatoid RCC (excluded by the lack of high-grade nuclear pleomorphism), mesenchymal tumors (rarely positive for cytokeratins), oncocytoma and chromophobe RCC (typically CD117-positive), and neuroendocrine neoplasms (excluded by negative chromogranin staining) [17,18].
Immunohistochemistry was decisive in establishing the diagnosis. Diffuse positivity for CK7, CK19, EMA, and AMACR supported MTSCC [18]. PAX8 confirmed renal origin, while CD117 and chromogranin were negative, excluding chromophobe/oncocytic and neuroendocrine tumors. The result of complete negativity for CD10 was helpful in ruling out papillary RCC [18]. The Ki-67 index was low (<10%), consistent with an indolent course [24].
In a series of nine MTSCC cases compared with ten papRCC cases, an immunohistochemical analysis showed 100% positivity for AMACR, CK7, and CK19 in MTSCC, but only 11% positivity for CD10 (versus 80% in papRCC), supporting CD10 as a useful discriminative marker [23]. Our case aligns with this pattern, showing MTSCC marker positivity and absent CD10 expression.
Genomic investigations further underline the distinction between MTSCC and papRCC. High-throughput sequencing and copy-number profiling have demonstrated recurrent chromosomal deletions together with mutations affecting genes such as NF2, CHEK2, and BRCA2 [16]. Characteristic molecular patterns include widespread losses (chromosomes 1, 4, 6, 8, 9, 13, 14, 15, and 22) in MTSCC, whereas papRCC more commonly displays gains of chromosomes 7 and 17 [13]. These chromosomal losses are observed even in high-grade or sarcomatoid cases, reinforcing the notion of MTSCC as a genetically distinct entity [5]. In advanced disease, alterations such as 1q gain and the homozygous deletion of CDKN2A/B have been linked to adverse outcomes [25,26]. The involvement of the MAPK and PI3K/AKT cascades has been proposed as a molecular basis for aggressive behavior [13,14,15,16], while Hippo pathway disruption—with bi-allelic inactivation of NF2, PTPN14, and SAV1 and increased nuclear expression of YAP1—has also been implicated as a potential oncogenic driver [27].
Taken together, MTSCC demonstrates a reproducible genomic profile, dominated by multiple chromosomal losses and lacking the gain patterns of papRCC. Additional alterations such as CDKN2A/B deletions and Hippo pathway dysregulation may underlie the rare aggressive phenotypes [5,13,16,25,26,27].
Clinically, most patients experience favorable outcomes after complete surgical excision [5,16]. However, sarcomatoid or high-grade variants are associated with a poor prognosis, often with a period of survival shorter than one year [15,17]. Because of their aggressive nature, sarcomatoid or high-grade MTSCC variants warrant more intensive surveillance compared with conventional cases [16,28]. In practical terms, this can involve CT or MRI performed every 3–6 months during the first two years, with annual imaging thereafter and follow-up extended beyond five years. Such a strategy is justified by the elevated risk of early relapse and metastatic spread [16,28]. Definitive management continues to rely on surgical resection, whereas systemic therapeutic approaches for advanced stages have not yet been standardized; recent reports suggest potential benefit from immune checkpoint blockade, particularly Ipilimumab plus Nivolumab, especially in cases with PD-L1 expression [15,29].
4. Conclusions
Mucinous tubular and spindle cell carcinoma is a rare and distinctive subtype of renal cell carcinoma. Accurate diagnosis requires the careful integration of clinical presentation, imaging, histopathological architecture, and immunohistochemical profile. Hallmark features—including a mucinous stroma, tubular and spindle cell arrangement, diffuse CK7/CK19/EMA/AMACR positivity, complete CD10 negativity, and a low Ki-67 proliferation index—are essential in distinguishing MTSCC from its histologic mimics.
Emerging evidence indicates that molecular profiling provides further insight to distinguish MTSCC from papillary RCC and recognize aggressive variants with less favorable outcomes. This biological heterogeneity emphasizes the importance of precise classification and long-term follow-up, even in low-grade cases.
We acknowledge that the present report is limited by its single-patient design and the absence of a comparative cohort, which inevitably restrict the generalizability of conclusions. Nevertheless, in this case, nephrectomy achieved complete disease control, with the patient remaining disease-free at five years, underscoring surgical excision as the cornerstone of therapy in localized disease and the necessity of prolonged surveillance.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Lopez-Beltran A. Scarpelli M. Montironi R. Kirkali Z. 2004 WHO classification of the renal tumors of the adults Eur. Urol.20064979880510.1016/j.eururo.2005.11.03516442207 · doi ↗ · pubmed ↗
- 2Moch H. Cubilla A.L. Humphrey P.A. Reuter V.E. Ulbright T.M. The 2016 WHO classification of tumours of the urinary system and male genital organs. Part A: Renal, penile, and testicular tumours Eur. Urol.2016709310510.1016/j.eururo.2016.02.02926935559 · doi ↗ · pubmed ↗
- 3WHO Classification of Tumours Editorial Board Urinary and Male Genital Tumours 5th ed.International Agency for Research on Cancer Lyon, France 2022978-92-832-4503-3
- 4Tsang S.L. Hsu S.S. An C. Ho S. Ng A. Mucinous Tubular and Spindle Cell Carcinoma: Case Report and Literature Review J. Kidney Cancer VHL 20251261110.15586/jkcvhl.v 12i 1.35440027351 PMC 11870379 · doi ↗ · pubmed ↗
- 5Hasan N. Moatasim A. Mucinous tubular and spindle cell carcinoma with high-grade transformation: Case report Surg. Exp. Pathol.20225410.1186/s 42047-022-00105-x · doi ↗
- 6Ling C. Tan R. Li J. Feng J. Mucinous tubular and spindle cell carcinoma of the kidney: A report of seven cases BMC Cancer 20232381510.1186/s 12885-023-11252-z 37649003 PMC 10470144 · doi ↗ · pubmed ↗
- 7Fine S.W. Argani P. De Marzo A.M. Delahunt B. Sebo T.J. Reuter V.E. Epstein J.I. Expanding the histologic spectrum of mucinous tubular and spindle cell carcinoma of the kidney Am. J. Surg. Pathol.2006301554156010.1097/01.pas.0000213271.15221.e 317122511 · doi ↗ · pubmed ↗
- 8Wang H. Peng X. Li L. Yang Y. A retrospective study of imaging characteristics of mucinous tubular and spindle cell carcinoma in the kidney Front. Oncol.202515151556910.3389/fonc.2025.151556940182031 PMC 11965112 · doi ↗ · pubmed ↗