PD-1 Expression Promotes Immune Evasion in B-ALL
Ana Casado-García, Gonzalo García-Aguilera, Julio Pozo, Ninad Oak, Susana Barrena, Belén Ruiz-Corzo, Jaanam Lalchandani, Ana Chamorro-Vera, Ana Castillo-Robleda, Beatriz Soriano, Silvia Alemán-Arteaga, Elena G. Sánchez, Jorge Martínez-Cano, Andrea López-Álvarez de Neyra

TL;DR
This study shows that PD-1 expression helps B-cell acute lymphoblastic leukemia (B-ALL) cells evade the immune system, and targeting PD-1 could offer new treatment options for childhood B-ALL.
Contribution
The study identifies PD-1 as a novel marker and therapeutic target for immune evasion in B-ALL.
Findings
PD-1 expression is upregulated in preleukemic cells and correlates with leukemia conversion in mice and humans.
PD-1 expression reduces antitumor immune responses but makes leukemic cells responsive to immune checkpoint blockade.
Targeting PD-1 restores NK cell-mediated tumor cell killing and eliminates tumor cells in mouse models.
Abstract
Background/Objectives: In children developing B-cell acute lymphoblastic leukemia (B-ALL), an immune evasion event takes place where otherwise “silent” preleukemic cells undergo a malignant transformation while escaping immune control, often through unknown mechanisms. Methods and Results: Here, we identify the upregulation of PD-1 expression in preleukemic cells, triggered by Pax5 inactivation in mice and correlating with the time of conversion to leukemia, as a novel marker that favors leukemia evasion. This increase in PD-1 expression is apparent across diverse molecular B-ALL subtypes, both in mice and humans. PD-1 is not required for B-cell leukemogenesis, but, in the absence of PD-1, tumor cells express NK cell inhibitory receptors, highlighting the necessity for leukemic cells to evade the host’s NK immune response in order to exit the bone marrow. PD-1 expression reduces natural…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
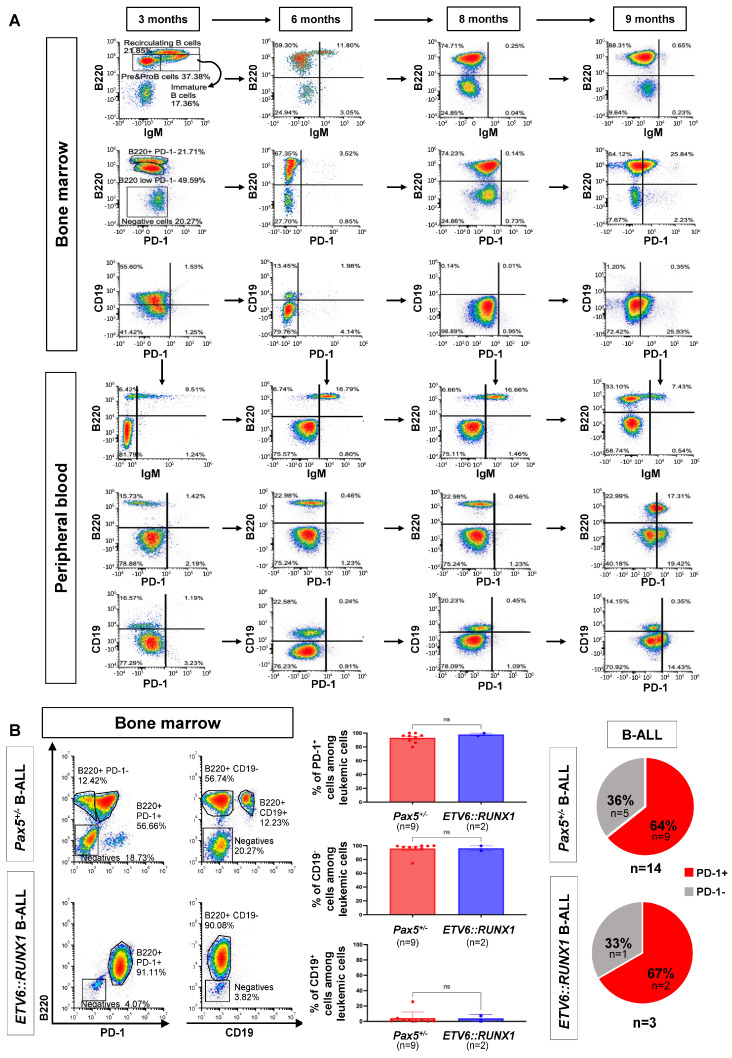 Figure 1
Figure 1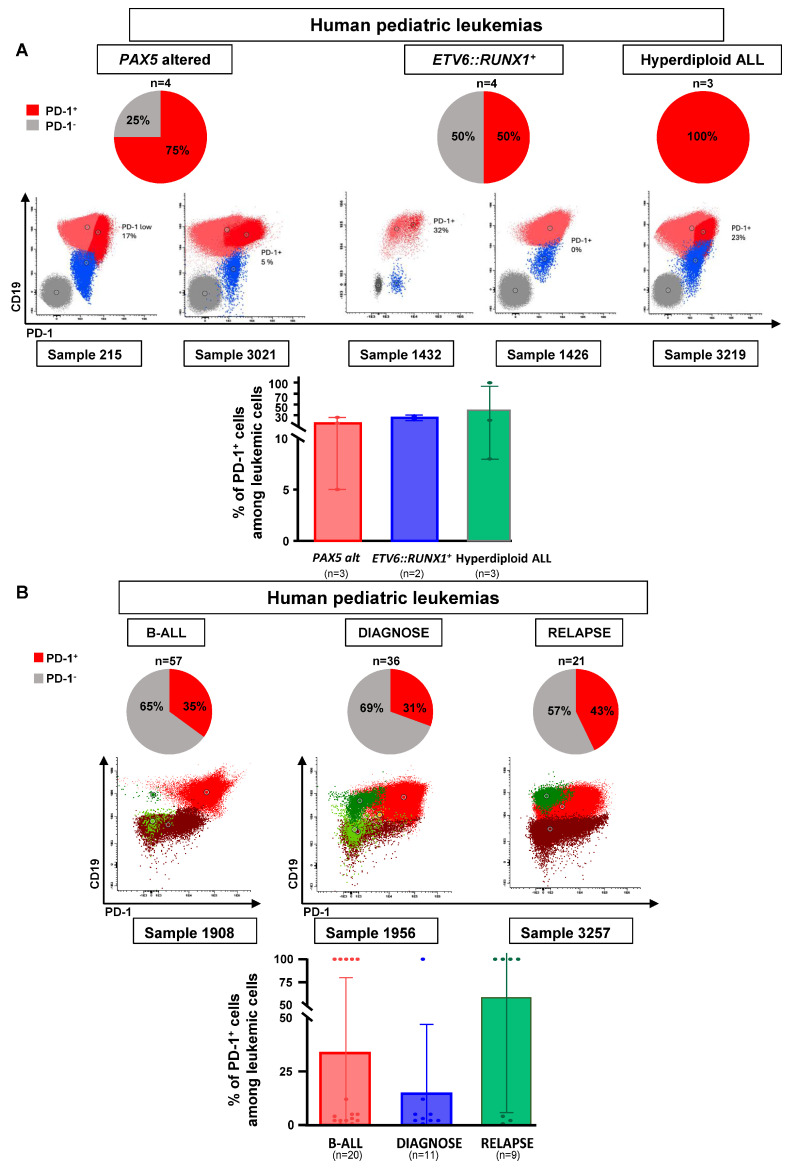 Figure 2
Figure 2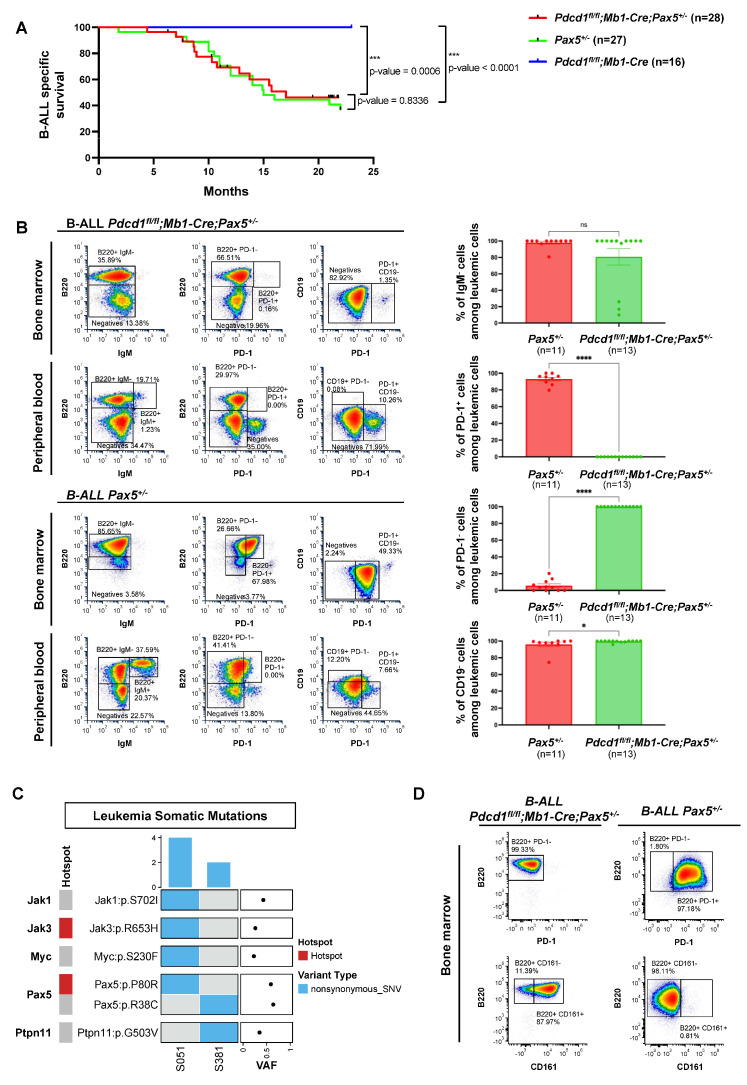 Figure 3
Figure 3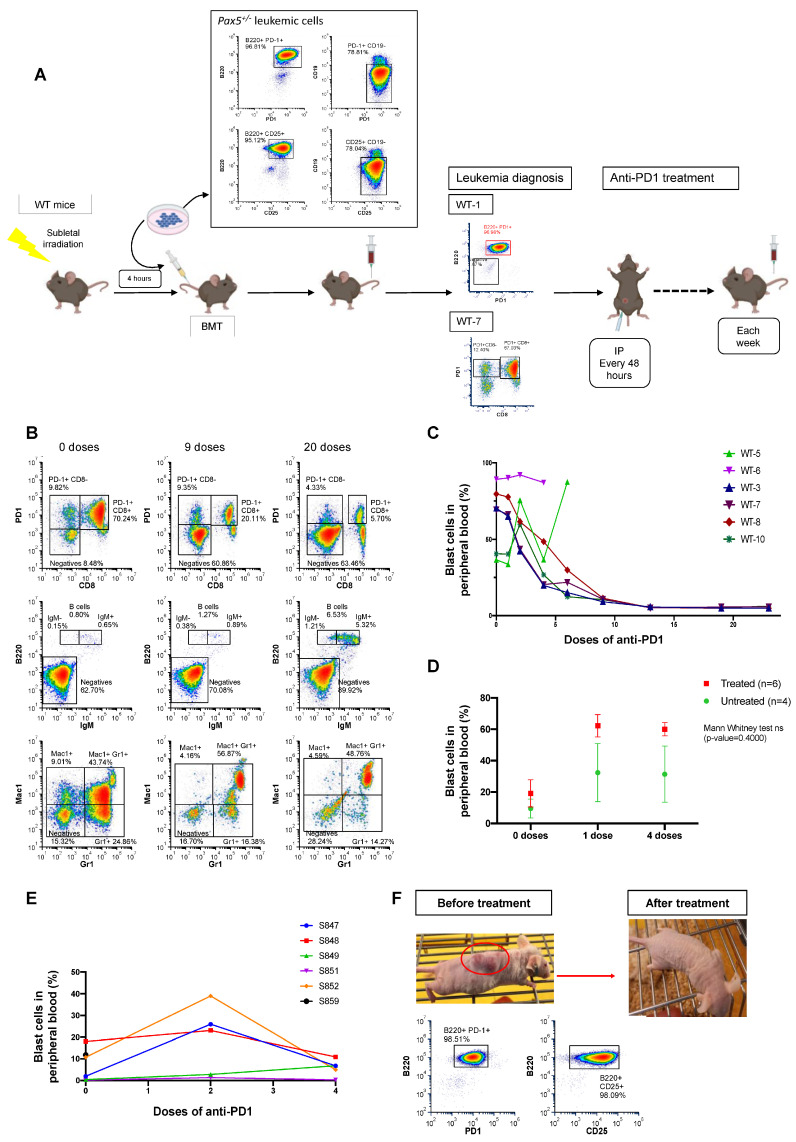 Figure 4
Figure 4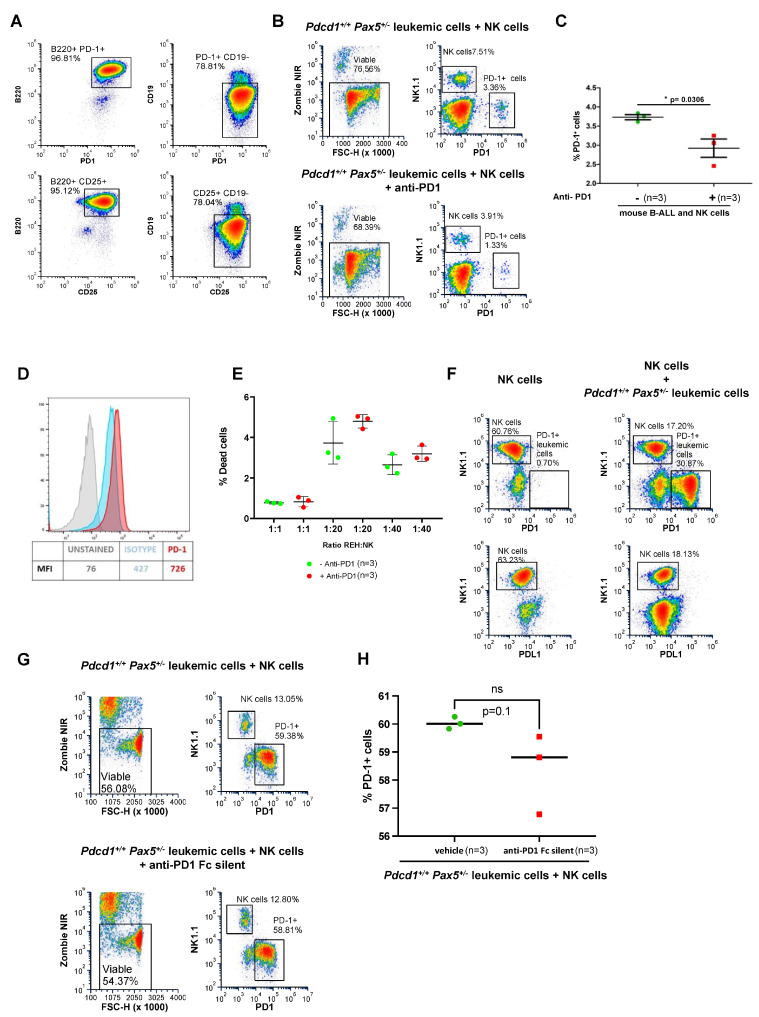 Figure 5
Figure 5- —MICIU/AEI/10.13039/501100011033
- —ERDF/EU
- —Fundación Síndrome de Wolf-Hirschhorn o 4p-“
- —Fundación Ramón Areces
- —Banco de Santander
- —Instituto de Salud Carlos III (ISCIII)
- —European Union (European Regional Development Fund (ERDF)/European Social Fund (ESF))
- —American Lebanese Syrian Associated Charities (ALSAC)
- —National Cancer Institute
- —National Institute of Allergy and Infectious Diseases
- —Histiocytosis Association
- —Cures within Reach
- —Asociación Pablo Ugarte
- —Junta de Castilla y León
- —Fundación Científica de la Asociación Española contra el Cáncer
- —TRANSCAN2023-1858-066–REACTION Project
- —FSE-Conserjería de Educación de la Junta de Castilla y León 2019 (ESF, European Social Fund)
- —Juan de la Cierva 2022
- —Ayuda para Contratos predoctorales para la formación de doctores
- —predoctoral fellowship FPI-UAM 2019
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAcute Lymphoblastic Leukemia research · CAR-T cell therapy research · Chronic Myeloid Leukemia Treatments
1. Introduction
In recent years, childhood B-lineage acute lymphoblastic leukemias (B-ALLs) have been extensively characterized at the genomic level [1,2,3,4,5]. Today, the vast majority of B-ALL cases can be categorized into distinct genetic subgroups, each associated with specific prognostic features and treatment responses [6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21]. In general, B-ALL is characterized by a very low mutational burden [2,5]. Furthermore, in 35–50% of all B-ALL cases, de novo genetic alterations involving genes encoding B-cell transcription factors serve as the primary oncogenic event, determining the disease’s biological and clinical attributes [22]. Similarly, pathogenic germline variants in genes encoding these same transcription factors, such as PAX5, predispose to B-ALL development [17,23,24]. However, these germline mutations, whether de novo or inherited, are not sufficient to trigger malignant transformations and require additional oncogenic secondary lesions to induce leukemia formation [14,17,20,22,25,26,27]. Notably, these secondary genetic events frequently also involve B-cell transcription factors and exhibit a remarkable specificity and recurrence pattern relative to the primary lesions.
Notably, alterations in PAX5, the gene encoding an essential protein required for B-cell development, are the most common central event in the process of B-cell leukemogenesis in children [1,14,17,20,22,28]. These PAX5 alterations, including focal deletions, single nucleotide variants and intragenic amplification, have been postulated to arrest lymphoid maturation, a feature characteristic of this disease. Nevertheless, their precise role in B-ALL development is not fully understood.
In this regard, it is well known that a decreased dosage of Pax5 activity significantly accelerates the development of precursor B-ALL in mice carrying additional fusion genes such as BCR::ABLp190 and ETV6::RUNX1 [29,30]. These findings suggest that the secondary PAX5 genetic alterations that accumulate during the process of malignant transformation might favor an escape from immune surveillance as B-ALL develops. Therefore, reinstating PAX5 function could potentially serve as a therapeutic approach to halt disease progression and even eliminate leukemic cells. Indeed, experimental evidence has corroborated this hypothesis, demonstrating that reintroducing endogenous Pax5 expression in Pax5-deficient leukemic B cells induces disease remission in murine models [31]. The initiation of Pax5 expression mediates B-cell commitment during normal hematopoietic differentiation [32], and its removal appears to be required to promote B-ALL development [33]. Although these results suggest that PAX5 downregulation plays a role in facilitating B-leukemogenesis, such an activity has yet to be directly demonstrated. Determining the mechanisms underlying leukemia formation is essential for improving B-ALL therapy and guiding approaches towards prevention [34,35].
The earliest stages of B-ALL development in children typically go undetected [34], making it nearly impossible to study the initial phases of leukemic transformation in humans. Thus, we have utilized the Pax5^+/−^ leukemia-prone model to investigate the mechanisms underlying immune evasion during progression to B-ALL. Mice heterozygous for Pax5, when exposed to infections, recapitulate the preleukemia-to-leukemia progression found in humans harboring the heterozygous germline PAX5 c.547G>A pathogenic variant [24,36,37,38,39]. This preleukemia is defined as an at-risk population of normal-appearing cells from which B-ALL develops. These cells have the capacity to undergo malignant transformation; however, leukemia only appears in a small fraction of predisposed carriers when they are exposed to certain environmental factors [34,40].
A substantial body of evidence underscores the significance of inhibitory receptors, commonly referred to as “immune checkpoints,” in tumor-mediated immune suppression. Among immune checkpoint proteins, Programmed Cell Death 1 (PD-1) is mainly expressed in activated T cells and macrophages, and, upon interaction with its ligand PD-L1 (Programmed Cell Death Ligand 1) expressed in tumor cells, PD-1 attenuates antitumor immune responses [41,42,43,44]. In this study, we show that B-ALL progression is associated with the upregulation of PD-1 in preleukemic cells and correlated with the time to malignant transformation in mice. This increase in PD-1 expression is apparent across diverse molecular B-ALL subtypes in mice and humans. Notably, targeting this B-ALL-associated PD-1 expression conferred clinical benefits by restoring NK-mediated tumor cell killing [45] in vitro and eliminating tumor cells in vivo in mice engrafted with B-ALL. These results identify PD-1 as a new therapeutic target in leukemic progression and provide new opportunities for the treatment or even prevention of childhood B-ALL.
2. Methods Details
2.1. Mouse Model for Natural Immune Stress-Driven Leukemia
Pax5^+/−^, Sca1-ETV6-RUNX1, Mb1-Cre and Pdcd1^fl/fl^ (C57BL/6-Pdcd1^tm1^.^1Mrl^; Taconic Model #13976) mice are as previously described [46,47,48]. Pax5^+/−^ mice were crossed with Pdcd1^fl/fl^ mice and Mb1-Cre animals to generate mice of the Pdcd1^fl/fl^;Mb1-Cre;Pax5^+/−^ genotype. Mice were bred and maintained under specific pathogen-free (SPF) conditions until exposed to conventional pathogens present in non-SPF animal facilities, as previously described [36,37,38,39,47,49]. Pdcd1^fl/fl^;Mb1-Cre;Pax5^+/−^ mice were treated with a cocktail of antibiotics (ampicillin, 1 g/L, Ratiopharm; vancomycin, 500 mg/L, Cell Pharm; ciprofloxacin, 200 mg/L, Bayer Vital; imipenem, 250 mg/L, MSD; metronidazole, 1 g/L, Fresenius) added to their drinking water ad libitum for a period of eight weeks, with the aim to facilitate leukemia development as previously described [37].
All mouse experiments were performed in accordance with the applicable Spanish and European legal regulations and had been previously authorized by the pertinent institutional committees of both the University of Salamanca and Spanish Research Council (CSIC). Both male and female mice were used in all the studies. Housing environmental conditions included a temperature of 21 °C ± 2 °C, humidity of 55% ± 10% and a 12 h:12 h light/dark cycle. Mice had access to food and water ad libitum. During housing, animals were monitored daily for health status. No data were excluded from the analyses. The inclusion criterion was based on the genotype of the mouse: transgenic versus control littermate. The investigator was not blinded to the group allocation.
Pax5^+/−^, Sca1-ETV6-RUNX1 and Pdcd1^fl/fl^;Mb1-Cre;Pax5^+/−^ mice of a mixed C57BL/6×CBA background were used in this study. For the relevant experiments, Pax5^+/−^, Sca1-ETV6-RUNX1 and Pdcd1^fl/fl^;Mb1-Cre;Pax5^+/−^ littermates were used. When animals showed signs of distress, they were humanely euthanized and their organs were harvested. All organs were macroscopically inspected under the stereomicroscope, and representative tissue samples were cut and immediately fixed and stained for subsequent histological analysis. Differences in Kaplan–Meier survival plots of transgenic and WT mice were analyzed using the log-rank (Mantel–Cox) test. Statistical analyses were performed by using GraphPad Prism v8.2.1 (GraphPad Software).
The B-ALL-specific survival curve of Pdcd1^fl/fl^;Mb1-Cre;Pax5^+/−^ mice (n = 28), Pax5^+/−^ mice (n = 27) and Pdcd1^fl/fl^;Mb1-Cre mice (n = 16) following exposure to common mouse pathogens was defined by the log-rank (Mantel–Cox) test. p-values are shown in the corresponding figures.
2.2. B-ALL Patient-Derived Xenograft (PDX) Models
Leukemic patient-derived xenografts (PDXs) were obtained from the St. Jude Children’s Research Hospital Public Resource of Patient-derived and Expanded Leukemias (PROPEL) (include https://propel.stjude.cloud (accessed on 22 February 2024)). Briefly, PDXs were established by the tail vein injection of primary human leukemia cells into 8–12-week-old sub-lethally irradiated (250 Rad) NOD.Cg-Prkdc^scid^ IL2rg^tm1Wjl^/SzJ (NSG) mice (The Jackson Laboratory_ RRID:IMSR_JAX:005557) [50]. Spleen cells harvested from engrafted mice were used for expansion in subsequent passages. The level of engraftment was monitored by monthly saphenous vein bleeds and flow cytometric analysis for human CD45^+^ and CD19^+^ cells.
2.3. Leukemic Pax5+/− Pro-B-Cell Culture
Iscove’s Modified Dulbecco’s Medium (IMDM- Thermo Fisher Scientific Inc. Alcobendas (Madrid), Spain) supplemented with 50 μmol/L β-mercaptoethanol, 1 mmol/L l-glutamine, 2% heat-inactivated FCS, 1 mmol/L penicillin–streptomycin (BioWhittaker- Merck Life Science S.L.U. Madrid 28006, Spain) and 0.03% (w/v) primatone RL (Sigma_ Merck Life Science S.L.U. Madrid 28006, Spain) was used for pro-B-cell culture experiments. Leukemic cells isolated by magnetic-activated cell sorting for B220^+^ (Milteny Biotec S.L. 28223 Pozuelo de Alarcón (Madrid) Spain) from BM were cultured on Mitomycin C-treated ST2 cells in IMDM without IL-7 (R&D Systems Bio-Techne R&D Systems, S.L.U. 28046 Madrid, Spain). The ST2 murine cell line, which is derived from mouse bone marrow and is used in research to promote the growth of haematopoietic cells, was obtained from Dr. Meinrad Busslinger [33].
2.4. Anti-PD1 Inhibitor In Vitro Experiments
Leukemic Pax5^+/−^ pro-B cells expressing PD-1 were seeded at 10^6^/3 mL/well in 6-well plates and treated with or without (vehicle) anti-PD1 monoclonal antibody (BioXCell; BE0146; 7.36 μg/mL) for 4, 8 or 24 h before being subjected to a cell viability assay, as described below. Leukemic Pax5^+/−^ pro-B cells expressing PD-1 were also treated with or without (vehicle) anti-PD1, which lacks the ability to bind to Fc receptors (Fc-silent RMP1-14) for 8 h before being subjected to a cell viability assay.
2.5. Viability Assays
Cells from anti-PD1 inhibitor cultures were washed twice with PBS (5′, 1000 rpm, 4 °C). Then, samples were stained with the fixable Zombie NIR viability dye kit (BioLegend; 423105_ Palex Medical S.A. Barcelona 08174-Spain) following the manufacturer’s instructions. Subsequently, cells were washed with PBS supplemented with 1% FCS to eliminate the excess of the viability marker (5′, 1400 rpm, RT). Samples were acquired in a Cytek Northern Light 2000 spectral cytometer (two lasers, red and blue) and analyzed with FCS Express 7 Plus software (version 7.08.0018). The specific fluorescence of the fluorophores and the known forward and orthogonal light scattering properties of mouse cells were used to establish gates. A total of 100,000 cells per sample were assessed. The difference between experimental variables was determined using the Mann–Whitney U test.
2.6. BM Transplantation Experiments
Leukemic Pax5^+/−^ pro-B cells expressing PD-1 were injected intravenously into sub-lethally irradiated (4 Gy) secondary recipient 8-week-old male syngeneic mice (C57BL/6 × CBA), NOD/SCID or nu/nu mice, respectively. Disease development in the recipient mice was monitored by periodic peripheral blood (PB) analysis until blast cells were detected. Then, mice were treated with anti-PD1 or placebo and assessed for B-ALL progression as indicated below. The study of PD-1 targeting as a potential therapeutic approach for childhood B-ALL was performed by using mouse PD-1^+^ leukemic pro-B cells injected through the tail vein of sub-lethally irradiated (4Gy) wild-type (n = 6) mice (C57BL/6 × CBA), NOD/SCID mice (n = 8) or nu/nu (n = 6) mice, respectively. No significant differences were observed in the decrease in the percentage of malignant cells between treated and untreated mice (Mann–Whitney test ns; p = 0.4000), showing the inefficacy of the anti-PD1 treatment in the absence of T and NK cells.
2.7. Preclinical Therapeutics
Anti-PD1 monoclonal antibody (BioXCell; BE0146_ Abyntek Biopharma, S.L. 48160 Derio—Bizkaia (Spain)) prepared in PBS was administered at 5 μg per mouse, intraperitoneally every 48 h, after animals had developed a detectable leukemic burden (percentage of blasts superior to 15% in PB), as documented by FACS analysis of PB. Disease progression in recipient mice was monitored by periodic PB analysis.
For a detailed description of methods, see the Supplemental Information.
3. Results
3.1. B-ALL Development Screens in Genetically Predisposed Mice Identify Cancer Cell-Autonomous Upregulation of the Inhibitory Molecule PD-1
In order to understand the sequential events leading to immune escape during B-ALL development, we used serial bone marrow aspirates, coupled with a sample analysis using spectral flow cytometry, in Pax5^+/−^ mice to characterize the immuno-regulatory events underlying the preleukemia-to-leukemia conversion (Figure 1). In human patients carrying inherited mutations of PAX5, preleukemic cells progress to B-ALL by losing the wild-type PAX5 allele due to a secondary structural aberration of chromosome 9p [24]. This is phenocopied in the B-ALLs developing in the Pax5^+/−^ mouse model [36,37,38,39,49], thus demonstrating the importance of the biallelic alterations of PAX5 for leukemia progression in this B-ALL subgroup. In these mice, upon the loss or marked reduction in Pax5 activity, the majority of B-ALLs lose CD19 expression, similar to what is observed in human PAX5-associated predisposition to childhood B-ALL [24]. Thus, surface CD19 expression is a very useful surrogate marker of Pax5 genetic status in mice and allows for the identification of potential immuno-regulatory factors that might correlate with leukemic conversion.
Using the aforementioned approaches, we first analyzed the expression of PD-1 (CD279) and PD-L1 (CD274) on Pax5^+/−^ preleukemic cells as they switched to a leukemic state following immune stress in the Pax5^+/−^ model [36,37,38,39,49]. Pax5^+/−^ preleukemic cells (B220^+^ IgM^−^) did not express PD-1 or PD-L1, and, in spite of their accumulation over time, they were retained in the bone marrow (BM) (Figure 1A, Figures S1 and S2). In contrast, PD-1 was upregulated upon the conversion to leukemic B cells in the BM, defined by the lack of CD19 expression in parallel with their emergence in the peripheral blood (PB) (Figure 1A and Figure S3A), indicating a loss of or marked reduction in Pax5 activity similar to what is found in human leukemia [24,36,37,38,39,49]. To examine whether a similar increase in PD-1 expression with leukemia evolution is observed in other mouse leukemia models, we evaluated, together with B-ALLs appearing in Pax5^+/−^ (n = 14) animals, those that originated in transgenic Sca1-ETV6-RUNX1 (n = 3) mice [36,47]. We found a detectable PD-1 expression in leukemic cells in 9 of the 14 (64%) Pax5^+/−^ B-ALL samples, and also in 2 of the 3 (67%) Sca1-ETV6-RUNX1^+^ B-ALL samples (Figure 1B and Table S1). All these mouse B-ALLs lacked surface CD19 expression, in agreement with the loss of or marked reduction in Pax5 activity, similar to what is found in human leukemia [24,36,37,38,39,49]. Thus, we next performed whole exome sequencing (WES) of paired tumor and germline samples from Pax5^+/−^ tumors to study the status of the wild-type Pax5 allele within PD-1+ leukemic cells (Figure S3B). WES identified four mutations at the Pax5 locus, as well as other leukemia hotspot mutations such as Nras:p.Q61H and Jak3:p.R653H. Of note, we could not assess the copy-number or structural alterations via WES. Recurrent genetic alterations affecting the wild-type Pax5 allele were detected within PD-1+ leukemic cells in some cases (Figure S3B). Importantly, no PD-1 expression was detected in normal precursor B cells or in CD19-expressing B-ALLs in either mouse model (Figure 1A,B), consistent with the genetic evidence showing that PD-1 expression is below detection in the presence of Pax5 [51], and that the expression of PD-1 is consistently upregulated in Pax5-deficient precursor B cells [51]. Taken together, these data illustrate that the genetic alterations affecting Pax5 that accumulate during the process of malignant transformation are associated with PD-1 upregulation upon leukemic conversion in genetically predisposed mice with acquired or germline alterations predisposing to B-ALL.
3.2. PD-1 Expression in Human B-ALL
To explore the clinical relevance of these results in humans, we subsequently investigated the expression of PD-1 in primary and xenografted human B-ALL samples, including those of the PAX5-alt subtype (where CD19 expression is maintained because the activation of the CD19 promoter in human B-ALL cells is not solely dependent on PAX5 activity [52]) and ETV6::RUNX1^+^ and hyperdiploid subtypes. PD-1 expression was present in 50–100% of these childhood B-ALL xenografts (n = 11) (75% in PAX5-alt B-ALLs (3 out of 4 B-ALLs), 50% in ETV6::RUNX1^+^ B-ALLs (2 out of 4 B-ALLs) and 100% in hyperdiploid B-ALLs (3 out of 3 B-ALLs)) (Figure 2A, Figures S4 and S5 and Table S2) and approximately in 31% of diagnostic (11 of 36 B-ALLs) and in 43% of relapsed (9 of 21 B-ALLs) childhood B-ALL primary samples (Figure 2B and Figure S6 and Table S3). However, it is important to note that, in human leukemias like in mice, PD-1 expression is not solely dependent on PAX5 activity (Table S2). In summary, PD-1 upregulation occurs in all molecular B-ALL subgroups tested, although the percentage of PD-1^+^ leukemic cells varied from 2 to 100% per sample. Since, until now, PD-1 antibodies are not usually included in most B-ALL marker and diagnostic panels, further prospective studies in a larger dataset of B-ALL cases are needed to confirm the findings linking leukemia cell genomics and PD-1 expression. Still, our observations are consistent with an “immunoediting” process, whereby intratumoral heterogeneity results in the selection of leukemic clones that can avoid elimination by the immune system and thus leave the bone marrow. These findings suggest that there may be a suppression of immune surveillance across human B-ALL subtypes.
3.3. PD-1 Is Not Required for Pax5-Dependent B-Cell Leukemogenesis
PD-1 expression has been recently reported as a marker of leukemic stem cells (LSCs) in T-ALL, and anti-PD1 treatment eliminates such LSCs in a cell-autonomous manner [53]. To investigate the role of PD-1 signaling in Pax5-dependent B-ALL, and to ascertain if, also in human B-ALLs, PD-1^+^ cells are functionally required for B-leukemogenesis, we used the PD-1 conditional knockout (Pdcd1^fl/fl^) mouse (Figure 3). Conditional Pdcd1^fl/fl^;Mb1-Cre;Pax5^+/−^ mice were generated, where Pdcd1 is deleted upon B-lineage commitment at the pro-B-cell stage, and we examined whether Pdcd1^fl/fl^;Mb1-Cre;Pax5^+/−^ animals were prone to infection-induced B-ALL. Pdcd1^fl/fl^;Mb1-Cre;Pax5^+/−^ mice were exposed to natural infections, and B-ALL development was monitored. We observed that B-ALL appeared in Pdcd1^fl/fl^;Mb1-Cre;Pax5^+/−^ mice, where 46% (13 out of 28 mice) developed the disease, closely resembling the incidence, latency and overall survival of Pax5^+/−^ animals (ns, p-value = 0.8336) (Figure 3A). This observation supports the notion that PD-1 is not required for B-leukemogenesis. The characterization of Pdcd1^fl/fl^;Mb1-Cre;Pax5^+/−^ B-ALLs showed that they are histologically and phenotypically similar to those appearing in Pax5^+/−^ mice (Figure 3B and Figure S7A–C).
To identify somatically acquired second hits leading to B-ALL development in Pdcd1^fl/fl^;Mb1-Cre;Pax5^+/−^ mice, we performed whole-genome sequencing of paired tumor and germline samples from two Pdcd1^fl/fl^;Mb1-Cre;Pax5^+/−^ tumors. We identified several somatically acquired recurrent mutations and copy-number variations involving B-cell transcription factors in diseased Pdcd1^fl/fl^;Mb1-Cre;Pax5^+/−^ mice (Figure S7D). To compensate for their inability to upregulate PD-1, the B-ALLs emerging in PD-1-deficient mice express the NK cell inhibitory receptor CD161, something that has never been previously reported in PD-1+ B-ALLs (Figure 3D and Figure S7E,F). Overall, the same genes appear mutated in Pax5^+/−^ murine leukemias [36,37,38,39,49] as in human B-ALL samples [2,5,17,54]. Thus, the drivers of B-ALL appear similar in Pdcd1^fl/fl^;Mb1-Cre;Pax5^+/−^ and Pax5^+/−^ mouse leukemic cells as well as human B-ALL blasts. However, in the absence of PD-1, tumor cells express the NK cell inhibitory receptor CD161, highlighting the importance of NK-mediated immune surveillance and the necessity for leukemic cells to evade the host’s NK immune response in order to exit the bone marrow. Future studies exploring cohorts of patients are required to better establish whether CD161 expression implies a new category of immunoevasive B-ALL subtypes.
3.4. Anti-PD1 Treatment Restores the Immune Capacity to Eliminate PD-1-Positive Tumor Cells in Mice Engrafted with B-ALL
Building on our findings, we next determined the biological consequences of PD-1 upregulation on antitumor immune response. Thus, we examined the potential of targeting PD-1 for B-ALL treatment (Figure 4). Here, in order to better understand the results from spectral cytometry, it is important to mention that, since one of the main functions of Pax5 is maintaining non-B-cell genes repressed during B-cell differentiation, B-ALL tumors arising in Pax5^+/−^ mice may mimic a Pax5^−/−^ phenotype and express promiscuous surface lineage markers such as CD8, Mac1 or Gr1 (Figure 4B). In contrast to what has been reported for T-ALL PD-1^+^ leukemia stem cells [53], in vitro PD-1 targeting did not induce the apoptosis of PD-1^+^ B-ALL cells (either of human or mouse origin) (Figure S8A,B). However, in vivo PD-1 targeting in B-ALL mice efficiently reduced disease burden and extended overall survival versus placebo-treated mice (p = 0.0177; Figure 4A–C and Figure S9). These findings suggest that PD-1 targeting released the immune-suppressive regulation and restored the tumor-specific cytotoxicity of either cytotoxic T lymphocytes or NK cells, which are the cellular components mediating the effects of the PD-1 blockade [55,56].
3.5. PD-1 Targeting Sensitizes PD-1-Positive B-ALLs to NK Cell-Mediated Killing
To determine whether the therapeutic effect of PD-1 targeting in B-ALL mice is dependent on T or NK cells, we repeated the checkpoint targeting strategy (Figure 4) in NOD/SCID mice, which lack B and T cells and have defective natural killer (NK) cell function. We recapitulated B-ALL disease in NOD/SCID mice by tail-vein injecting the PD-1^+^ leukemic B cells. To dissect the impact of PD-1 targeting in leukemic NOD/SCID mice, we initiated the treatment once blast cells were detected in PB. Unlike in immunocompetent animals, PD-1 targeting in PD-1^+^ B-ALL NOD/SCID mice did not reduce disease burden (Figure 4D). This similar in vivo growth of tumor cells in NOD/SCID mice excluded a tumor cell-intrinsic effect of PD-1-targeting antibodies. However, when we similarly recapitulated PD-1^+^ B-ALL disease in nu/nu mice, which lack T cells but possess NK cells, PD-1 targeting efficiently reduced the disease burden (Figure 4E,F). These findings are consistent with previous observations suggesting a T-cell independent immunosurveillance mechanism in the conversion of the preleukemic clone into a full-blown B-ALL [34,37,57]. To better understand how B-ALLs can escape NK cell surveillance through the PD-1 checkpoint, we performed ex vivo NK cytotoxicity assays using both mouse (S748 cells) and human B-ALL (REH cells). NK cytotoxicity assays showed that the targeting of PD-1 using the specific anti-PD1 antibody sensitizes both mouse and human B-ALLs to NK cell-mediated killing (Figure 5A–E). Taken together, our results indicate that PD-1 is central to mediating the in vivo escape of B-ALL cells from surveillance by NK cells, and that PD-1 targeting conferred clinical benefits by restoring NK-mediated tumor cell killing in vitro and eliminating tumor cells in vivo in mice engrafted with B-ALL.
3.6. PD-1 Directly Inhibits the Antitumor Activity of NK Cells on B-ALL
It has been proposed that the acquisition of PD-1 in the membrane via trogocytosis can inhibit NK cell response against tumor cells expressing PD-1 [58]. To test this hypothesis, we initially used PD-1^+^ S748 cells, which derive from the transformation of murine Pax5^+/−^ B cells under immune stress [36]; these S748 cells express high levels of PD-1 (Figure 5A), and we have used them extensively in previous studies [36]. We now co-cultured PD-1^+^ S748 leukemic cells together with NK cells. In the absence of tumor cells, NK cells did not stain for PD-1 (Figure 5F). Similarly, NK cells did not stain positively for PD-1 when incubated with PD-1^+^ S748 leukemic cells (Figure 5F), indicating that PD-1 was not endogenously expressed by NK cells and was not acquired from leukemic cells in these settings. Likewise, co-culturing preleukemic (i.e., PD-1-negative) Pax5^+/−^ cells with NK cells leads to cytotoxicity that is not affected by treatment with anti-PD-1 antibodies (Figure S10). Finally, to examine whether the therapeutic effect of PD-1 antibodies was due to antibody-dependent cellular cytotoxicity (ADCC), likely mediated by NK cells against PD-1^+^ leukemic cells coated with anti-PD1 antibodies, we used an engineered version of anti-PD-1 that lacks the ability to bind to Fc receptors (Fc-silent RMP1-14) [59]. A treatment with Fc-silent anti-PD1 antibodies did not affect the growth of PD-1^+^ S748 leukemic cells (Figure 5G,H), indicating that the therapeutic effect of anti-PD-1 antibodies was due to ADCC. Together, these results indicate that PD-1 expression by leukemic cells suppresses NK cell-mediated antitumor activity, facilitating immune evasion, bone marrow exit and disease dissemination. Importantly, this process can be prevented by treatment with PD-1-targeting antibodies. However, it cannot be discarded that there could be another mechanistic explanation for the protective function of PD-1 on leukemia cells, like, for example, the modulation of activating/inhibitory NK receptors.
4. Discussion
If childhood B-cell acute lymphoblastic leukemia (B-ALL) happens only in genetically predisposed individuals and is triggered by immune stress, then it is very likely a preventable disease. Therefore, it becomes imperative to delineate the processes implicated in leukemic progression, as these processes may provide windows of opportunity to intervene by stopping B-ALL in its tracks.
Here, our studies have uncovered a previously unknown molecular process that takes place during the initial stages of childhood B-cell leukemogenesis, allowing us to discover a readout of PAX5 inactivation, the most frequently altered transcription factor in B-ALL [1,14,17,20,22,28]. We demonstrate that the Pax5 genetic alterations that accumulate during the process of malignant transformation are associated with PD-1 upregulation upon the time of conversion to B-ALL in mice. PD-1 is upregulated in murine Pax5-deficient B-ALL because PD-1 is a target gene repressed by PAX5 in normal conditions [51]. This increase in PD-1 expression is also apparent across diverse molecular B-ALL subtypes in human B-ALL samples, although in mouse and human leukemias PD-1 expression is not solely dependent on PAX5 activity. However, it is not known whether PD-1 upregulation in Pax5-deficient B-ALL is accompanied by changes in membrane microdomain organization. This increase in PD-1 expression is also apparent across diverse molecular B-ALL subtypes in human B-ALL samples, although in mouse and human leukemias PD-1 expression is not solely dependent on PAX5 activity, and further studies should help to identify the mechanisms involved. In addition, PD-1 upregulation protects B-ALL blasts from NK-mediated immune surveillance. Conversely to what was observed in T-ALL [53], PD-1 is not functionally required for B-leukemogenesis. However, in the absence of PD-1, leukemic cells upregulate the NK cell inhibitory receptor CD161, underscoring the critical role of NK cell-mediated immune surveillance. This suggests that the evasion of NK cell-mediated immunity is essential for leukemic cells to exit the bone marrow. The importance of shielding B-ALL blasts from NK surveillance is further reinforced by the upregulation of alternative NK cell inhibitory molecules in B-ALLs arising in PD-1-deficient mice. Finally, our findings reveal that PD-1 targeting can serve as a therapeutic strategy to promote the NK cell-mediated killing of leukemia cells, providing new opportunities for the treatment or prevention of childhood B-ALL. Our findings could also justify the use of small molecular PD-1 inhibitors for oral administration in B-ALL patients with PD-1-expressing leukemic cells, currently in development for some solid tumors [60]. Nevertheless, the use of single agents against cancer is not very prudent and, therefore, combining PD-1 blockades with standard drugs could help in managing B-ALL better. However, this anti-PD-1 therapy could fail in clinical settings through the acquisition of other checkpoint inhibitors, and it can be associated with potential risks, like immunosuppression, autoimmunity, etc., that should be minimized by the early identification and early onset of a prophylactic treatment.
It is important to underscore that the identification of a biomarker correlating with environmental exposures along the trajectory of leukemic transformation has been achieved using predisposed mouse models where the B-ALL disease emerges naturally. This is relevant because many of the observations obtained previously using these mouse models have later been mirrored in pediatric B-ALL patients, as illustrated by the case where the B-cell alterations found in preleukemic Pax5^+/−^ mice [36] were later confirmed in children carrying PAX5 germline variants [61], or by the discovery that B-ALL driver genes are not targeted by AID in mice, subsequently validated in human ALL blasts [49], or by the identification of gut microbiome immaturity in B-ALL-predisposed mice [37], which was also later corroborated in children with B-ALL [62]. Hence, these preclinical mouse models, where B-ALL occurs naturally, are indispensable for elucidating the early phases of B-ALL development, which are typically unnoticed in children [34], making it nearly impossible to study the initial phases of leukemic transformation in humans. Likewise, recently it has been shown that NK cell-mediated cytotoxicity shapes the clonal evolution of human B-cell leukemia by showing that tumor cells are actively edited by NK cells during the equilibrium phase and use different avenues to escape NK cell-mediated eradication [63].
5. Conclusions
Overall, our study demonstrates that certain cases of human B-ALL express PD-1, and that targeting this pathway with checkpoint inhibitors can activate NK cell activity, thereby presenting a potential therapeutic opportunity. In fact, our data show that sustained leukemia suppression can be achieved with anti-PD1 single-agent treatment in the presence of a functional NK compartment. Accordingly, these findings form the foundation for a potential new approach to treat B-ALL, the commonest form of pediatric cancer and the leading cause of cancer-related death in children. These results are also of immediate diagnostic importance because they suggest that most PD-1-positive B-ALL cases may be rapidly detected using flow cytometry immunophenotyping to further guide classification and tailored therapy. Future studies exploring larger cohorts of patients will be required to better establish how PD-1 upregulation may be used to direct the daily clinical care of children with B-ALL.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Mullighan C.G. Goorha S. Radtke I. Miller C.B. Coustan-Smith E. Dalton J.D. Girtman K. Mathew S. Ma J. Pounds S.B. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia Nature 200744675876410.1038/nature 0569017344859 · doi ↗ · pubmed ↗
- 2Grobner S.N. Worst B.C. Weischenfeldt J. Buchhalter I. Kleinheinz K. Rudneva V.A. Johann P.D. Balasubramanian G.P. Segura-Wang M. Brabetz S. The landscape of genomic alterations across childhood cancers Nature 201855532132710.1038/nature 2548029489754 · doi ↗ · pubmed ↗
- 3Holmfeldt L. Wei L. Diaz-Flores E. Walsh M. Zhang J. Ding L. Payne-Turner D. Churchman M. Andersson A. Chen S.C. The genomic landscape of hypodiploid acute lymphoblastic leukemia Nat. Genet.20134524225210.1038/ng.253223334668 PMC 3919793 · doi ↗ · pubmed ↗
- 4Iacobucci I. Mullighan C.G. Genetic Basis of Acute Lymphoblastic Leukemia J. Clin. Oncol.20173597598310.1200/JCO.2016.70.783628297628 PMC 5455679 · doi ↗ · pubmed ↗
- 5Ma X. Liu Y. Alexandrov L.B. Edmonson M.N. Gawad C. Zhou X. Li Y. Rusch M.C. Easton J. Huether R. Pan-cancer genome and transcriptome analyses of 1699 paediatric leukaemias and solid tumours Nature 201855537137610.1038/nature 2579529489755 PMC 5854542 · doi ↗ · pubmed ↗
- 6Roberts K.G. Morin R.D. Zhang J. Hirst M. Zhao Y. Su X. Chen S.C. Payne-Turner D. Churchman M.L. Harvey R.C. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia Cancer Cell 20122215316610.1016/j.ccr.2012.06.00522897847 PMC 3422513 · doi ↗ · pubmed ↗
- 7Gu Z. Churchman M. Roberts K. Li Y. Liu Y. Harvey R.C. Mc Castlain K. Reshmi S.C. Payne-Turner D. Iacobucci I. Genomic analyses identify recurrent MEF 2D fusions in acute lymphoblastic leukaemia Nat. Commun.201671333110.1038/ncomms 1333127824051 PMC 5105166 · doi ↗ · pubmed ↗
- 8Iacobucci I. Li Y. Roberts K.G. Dobson S.M. Kim J.C. Payne-Turner D. Harvey R.C. Valentine M. Mc Castlain K. Easton J. Truncating Erythropoietin Receptor Rearrangements in Acute Lymphoblastic Leukemia Cancer Cell 20162918620010.1016/j.ccell.2015.12.01326859458 PMC 4750652 · doi ↗ · pubmed ↗