A Chromosomally Integrated T7 RNA Polymerase Enables T7-Derived Expression in Salmonella enterica without Compromising Virulence
Seungwoo Baek, Seoyeon Kim, Eun-Jin Lee

TL;DR
Scientists created a Salmonella strain that can use the T7 RNA polymerase system for protein expression without losing its ability to cause disease.
Contribution
A T7 RNA polymerase system was chromosomally integrated into Salmonella enterica without affecting its virulence or growth.
Findings
Salmonella-T7 supports robust, inducible expression of heterologous proteins using T7 promoters.
The engineered strain shows growth and virulence comparable to wild-type Salmonella.
The system enables T7-based expression in a pathogenic context for synthetic biology and vaccine studies.
Abstract
The T7 RNA polymerase (T7 RNAP) system has revolutionized protein expression in Escherichia coli due to its high transcriptional activity and tight regulation. However, Salmonella enterica, despite its close genetic similarity to E. coli, lacks a T7 RNAP system, limiting the use of T7-based vectors and tools in this pathogen. Establishing a T7-compatible Salmonella strain would enable the seamless application of E. coli-optimized expression systems for studies in a pathogenic context. We engineered S. enterica serovar Typhimurium strain 14028s to stably express T7 RNAP from the chromosome under the control of the lac promoter using the pGRG36 transposon system. The resulting strain (Salmonella-T7) supports robust IPTG-inducible expression of heterologous proteins from T7 promoter-driven vectors, such as the pET series. Salmonella-T7 exhibited growth kinetics comparable to wild-type…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
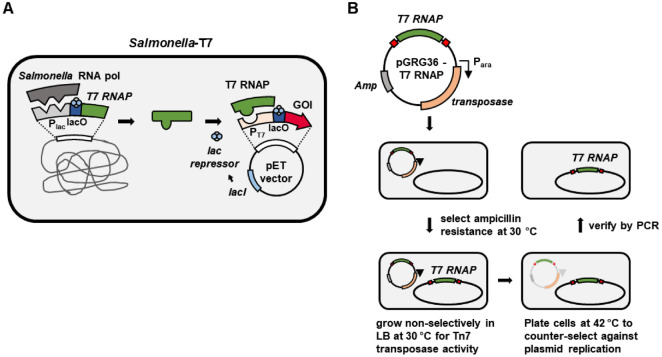 Figure 1
Figure 1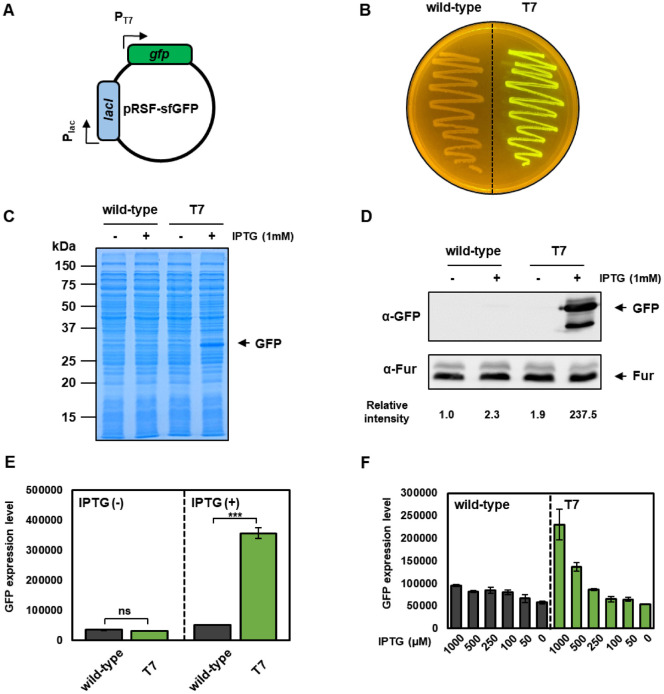 Figure 2
Figure 2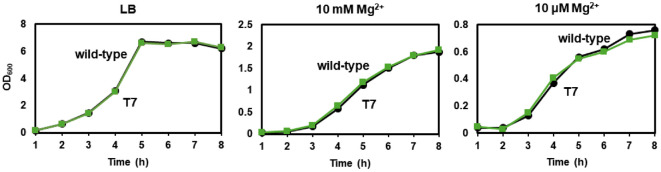 Figure 3
Figure 3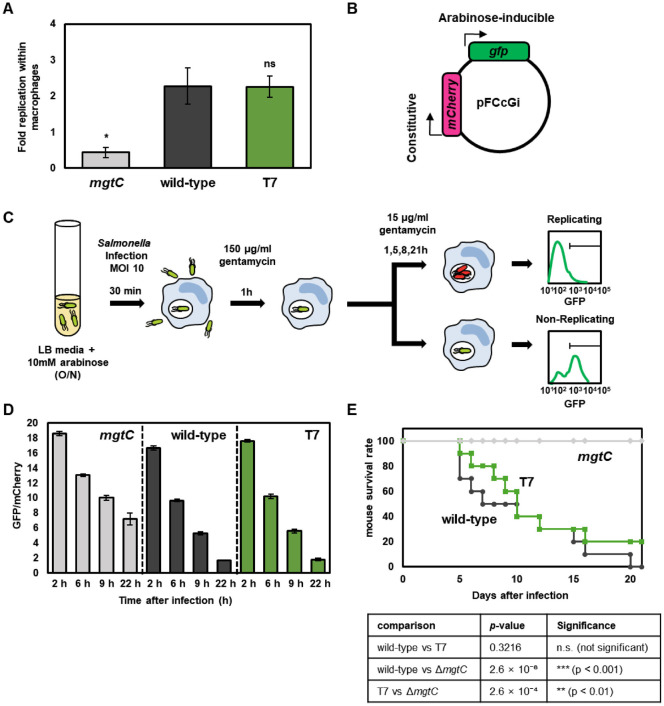 Figure 4
Figure 4 Figure 5
Figure 5- —National Research Foundation of Koreahttp://dx.doi.org/10.13039/501100003725
- —Korea University10.13039/501100002642
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacterial Genetics and Biotechnology · Salmonella and Campylobacter epidemiology · Escherichia coli research studies
Introduction
T7 RNA polymerase/promoter system has revolutionized protein production in Escherichia coli due to its exceptional transcriptional activity and tight regulation [1?-3]. Therefore, vectors such as the pET series, which exploit T7 promoters, are extensively applied in molecular biology and biotechnology for efficient expression of recombinant proteins [4???-8]. Yet, purification of heterologous proteins expressed in E. coli inherently results in their dissociation from their native physiological conditions [9], thereby losing endogenous modifications and regulatory influences. For example, even though Salmonella is closely related to E. coli in terms of phylogeny, expression of orthologous Salmonella genes in E. coli is often inefficient due to differences in regulatory and folding contexts, leading to poor protein yield and functionality [10].
To investigate proteins within their native context, researchers often rely on ectopic expression in the native host, typically by inserting regulated promoters such as the lac promoter (plac) in the chromosome. A two-part broad-host-range (BHR) plasmid system for T7 expression has also been developed [11]. While such systems can drive expression, they have limitations. Protein yields in the native host are often low because chromosomal expression systems fail to achieve sufficient expression levels. In single-vector expression systems, leaky expression often prevents tight regulation of the target protein. Although bipartite systems employing two plasmids can achieve tighter control, they require dual antibiotic selection, which is not ideal for studies involving pathogenic bacteria and their virulence. In Salmonella enterica, a widely used model organism for studying host-pathogen interactions [12, 13], it has been shown previously that heterologous expression under the control of the lac promoter compromises Salmonella virulence [14, 15].
In this study, we aimed to address these limitations by integrating T7 RNA polymerase into the chromosome of S. enterica. This engineered strain, hereafter referred to as Salmonella-T7, was designed to allow seamless use of existing T7-based vectors, such as the pET series, in a pathogenic Salmonella background. We demonstrate that Salmonella-T7 supports robust IPTG-inducible expression of heterologous proteins from pET vectors. Furthermore, because maintaining pathogenicity is essential for studying host-pathogen interactions, we evaluated whether this modification affects bacterial virulence. Our results show that Salmonella-T7 retains full virulence in macrophage infection assays and mouse models, establishing it as a powerful platform for both in vitro and in vivo studies of Salmonella physiology and heterologous protein expression in a pathogenic context.
Materials and Methods
Bacterial Strains, Oligonucleotides, and Growth Conditions
Bacterial strains, plasmids and oligonucleotides used in this study are listed in Table 1. All Salmonella enterica serovar Typhimurium strains are derived from the wild-type strain 14028s [16]. Bacteria were grown at 37°C in Luria-Bertani broth (LB), or in N-minimal medium (pH 7.7) [17] supplemented with 0.1% casamino acids, 38 mM glycerol, and the indicated concentrations of MgCl_2_. Escherichia coli DH5α was used as the host for plasmid DNA preparation. Antibiotics were used at the following concentrations: ampicillin, 50 mg/ml; gentamicin, 10 μg/ml; and kanamycin, 50 μg/ml.
Plasmid Construction
For insertion of the T7 RNA polymerase gene into the Salmonella chromosome, the plasmid pGRG36-T7 RNAP was constructed as follows. DNA fragments corresponding to the T7 RNA polymerase gene were amplified by PCR using the primer pair SPU284/SPU285 and plasmid pJLG038 [11] as the template. After purification, the PCR products were digested with XmaI and XhoI and cloned into the pGRG36 plasmid digested with the same enzymes. As the pGRG36 transposon system exhibits leaky expression, all cloning was performed in the presence of 0.2% glucose.
Measurement of Bacterial Growth
Growth of Salmonella containing chromosomally integrated T7 RNAP was measured. As a control, the wild-type Salmonella 14028s strain was used. For growth tests, strains were streaked onto solid LB plates and incubated at 37°C. A single colony was then inoculated into 2 ml of LB or N-minimal medium containing 10 mM Mg^2+^ and incubated at 37°C. A 1/100 dilution of pre-cultured samples was inoculated into 15 ml LB or N-minimal medium and cultured with shaking at 37°C. For growth in N-minimal medium containing 10 μM Mg^2+^, pre-cultured samples grown in N-minimal medium were washed twice with N-minimal medium without Mg^2+^ before inoculation. Then, the OD_600_ value was measured every hour.
Protein analysis by SDS-PAGE and immunoblotting
Overnight cultures were diluted 1:100 into 10 ml of LB medium with or without 1 mM IPTG and grown for 3 h. Cells were normalized by measuring OD_600_. Crude extracts were prepared in TBS (Tris-buffered saline) buffer by sonication, electrophoresed on 12% SDS-polyacrylamide gels. For Coomassie staining, the gel was stained with EZ-gel staining solution (DoGenBio: DG-GS1000, Korea) for 1 h at room temperature.
For Western blot analysis, proteins were transferred to nitrocellulose membranes after electrophoresis. Fur and GFP were detected using anti-Fur polyclonal and anti-GFP monoclonal antibodies (Roche: 1814460001, Switzerland), respectively. The blots were incubated with above antibodies overnight at 4°C, followed by incubation with horseradish peroxidase–conjugated anti-rabbit (Thermo Fisher Scientific: 31460, USA) or anti-mouse IgG (Sigma-Aldrich: NA931V, USA) secondary antibodies (1:10,000 dilution) for 1 h. Signals were detected using SuperSignal^®^ West Femto Maximum Sensitivity Substrate (ThermoFisher: 34095). The data are representative of at least two independent experiments, which gave similar results.
Quantification of GFP Fluorescence Using a Plate Reader
Overnight bacterial cultures were diluted 1:100 into 10 ml of N-minimal medium containing 0.01 mM Mg²+ and grown for 3 h. 1 mM of IPTG was then added, and cultures were incubated for an additional hour. For measuring GFP expression, cells were aliquoted into a 96-well black/clear-bottom plate (ThermoFisher), GFP fluorescence and OD_600_ were determined using a Synergy H1 plate reader (BioTek Instruments, USA) by measuring fluorescence at 485 nm excitation and 535 nm emission (GFP) and absorbance at 600 nm (OD_600_). GFP expression levels were calculated by dividing the GFP fluorescence values by the OD_600_.
Flow Cytometry
Macrophage-like J774A.1 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) at 37°C in a 5% CO_2_ atmosphere. Prior to infection, 5 × 10^5^ cells were seeded into 24-well plates and incubated for 20 h. Salmonella strains harboring either pFCcGi (a dual-fluorescence reporter plasmid expressing constitutive mCherry and inducible GFP) were grown overnight in LB medium. Bacterial cultures were used to infect macrophages at a multiplicity of infection (MOI) of 10. Plates were centrifuged at 1,500 rpm for 5 min at room temperature and incubated for 30 min to facilitate bacterial uptake. Extracellular bacteria were removed by washing three times with phosphate-buffered saline (PBS) and subsequently killed by incubating cells in DMEM supplemented with 10% FBS and 120 μg/ml gentamicin for 1 h. The medium was then replaced with fresh DMEM containing 12 μg/ml gentamicin, and infection was continued at 37°C. At indicated time points, macrophages were washed and lysed with PBS containing 0.1% Triton X-100. Intracellular bacterial fluorescence was assessed using a NovoCyte flow cytometer (ACEA Biosciences, USA). mCherry and GFP signals were excited at 488 nm, with emissions collected at 615 nm (red) and 530 nm (green), respectively. Data acquisition and analysis were performed using NovoExpress software (ACEA Biosciences) [18].
Macrophage Survival Assay
Intracellular survival assays were performed using the J774A.1 murine macrophage-like cell line as previously mentioned [19]. A total of 5 × 105 macrophages were seeded in 24-well plates containing Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and incubated at 37°C with 5% CO_2_. Overnight-grown bacteria were added to the macrophages at a multiplicity of infection (MOI) of 10. Plates were centrifuged at 1,500 rpm for 5 min at room temperature and incubated for an additional 30 min to facilitate bacterial uptake. Extracellular bacteria were removed by washing wells twice with phosphate-buffered saline (PBS), followed by incubation in DMEM supplemented with 10% FBS and 120 μg/ml gentamicin for 1 h to kill remaining extracellular bacteria. To determine intracellular bacterial counts at 1 h post-infection, macrophages were lysed with PBS containing 0.1% Triton X-100, and serial dilutions of the lysates were plated on Luria–Bertani (LB) agar. For 22 h time points, after the initial 1 h gentamicin treatment, the medium was replaced with fresh DMEM containing 12 μg/ml gentamicin, and incubation continued at 37°C. At 22 h, cells were lysed as described above, and viable intracellular bacteria were quantified by plating. Percentage survival was calculated by dividing colony-forming units (CFUs) recovered at 22 h by those recovered at 1 h. All experiments were performed in duplicate and repeated independently at least three times.
Mouse Virulence Assay
Six- to eight-week-old female C3H/HeN mice were inoculated intraperitoneally with approximately 10^3^ colony-forming units (CFU) of Salmonella strains. Mouse survival was monitored for 21 days. Virulence assays were repeated two times with consistent outcomes, and the presented data represent groups of ten mice per strain. All animals were housed in a temperature- and humidity-controlled facility with a 12 h light/12 h dark cycle. Experimental procedures were approved by the Korea University Institutional Animal Care and Use Committee (KUIACUC-2024-0054).
Results
Integration of T7 RNA Polymerase into the Salmonella Chromosome by Tn7 Transposon
To enable the use of T7 promoter-driven expression systems in S. enterica subsp. enterica serovar Typhimurium strain 14028s, we engineered a strain with chromosomally integrated T7 RNA polymerase under the control of the lac promoter and operator (plac/lacO), allowing IPTG-inducible expression (Fig. 1A). This strategy replicates the core regulatory logic of the E. coli BL21(DE3) system but in a pathogenic Salmonella background. The integration was performed using the pGRG36-T7 RNAP plasmid, which carries the T7 RNA polymerase gene and a temperature-sensitive replication origin (Fig. 1B) [20]. Transformation of wild-type Salmonella Typhimurium 14028s with this plasmid and selection at 30°C for ampicillin resistance induced expression of the Tn7 transposases, resulting in recombination of the T7 RNA polymerase gene into the Salmonella chromosome. Non-selective growth at 30°C facilitated site-specific insertion of the T7 RNA polymerase gene at the attTn7 site downstream of glmS. Counter-selection at 42°C eliminated the temperature-sensitive plasmid, and chromosomal integration was confirmed by PCR and sequencing. This genetic engineering approach generated a strain (Salmonella-T7) capable of expressing T7 RNA polymerase without the need for plasmid maintenance or antibiotic selection, providing a stable system for T7 promoter-driven expression in Salmonella.
T7 RNA Polymerase in Salmonella Tightly Regulates IPTG-Inducible GFP Expression
We next assessed whether the integrated T7 RNA polymerase is functional and capable of driving expression from T7 promoters. The pRSF-sfGFP reporter plasmid, a pET-based vector containing GFP under T7 promoter control [21], was introduced into both Salmonella-T7 and wild-type strains (Fig. 2A). When streaked onto LB agar plates containing 1 mM IPTG, only Salmonella-T7 colonies exhibited bright green fluorescence visible under UV illumination, while wild-type colonies remained non-fluorescent (Fig. 2B).
To further quantify expression, cultures were grown in LB broth in the presence or absence of 1 mM IPTG, and total protein extracts were analyzed by SDS-PAGE and Western blotting. In Coomassie-stained gels, a strong band corresponding to GFP (~27 kDa) was detected in Salmonella-T7 samples with IPTG addition but not in uninduced or wild-type samples (Fig. 2C). Western blotting using anti-GFP antibodies confirmed robust GFP production in Salmonella-T7 following IPTG induction (Fig. 2D). These results indicate that expression is tightly regulated, with minimal background in the absence of IPTG.
Fluorescence measurements in N-minimal medium supplemented with 10 mM Mg^2+^ supported these findings. GFP expression in Salmonella-T7 increased approximately 10-fold upon IPTG induction, while wild-type strains showed no significant change (Fig. 2E). Dose-dependent GFP expression demonstrated that Salmonella-T7 supports tunable GFP expression across IPTG concentrations from 50 μM to 1 mM (Fig. 2F). Collectively, these results establish that chromosomally expressed T7 RNA polymerase in Salmonella-T7 enables strong and tightly controlled expression from T7 promoters.
Insertion of T7 RNA Polymerase Does Not Affect Bacterial Growth
To determine whether chromosomal integration of T7 RNA polymerase impacts bacterial fitness, we compared the growth of Salmonella-T7 and wild-type 14028s in various culture conditions. Growth curves in LB medium, a rich medium for Salmonella, demonstrated comparable growth between the two strains, with OD_600_ values reaching stationary phase at approximately the same time (Fig. 3, left panel). To assess growth under nutrient-limiting conditions, strains were cultured in N-minimal medium supplemented with either 10 mM or 10 μM Mg^2+^. Again, no significant differences in growth rates or final cell densities were observed between Salmonella-T7 and the wild-type (Fig. 3, center and right panels). Together, these results indicate that chromosomally expressed T7 RNA polymerase does not impose a detectable metabolic burden or growth defect in either rich or minimal media.
Salmonella-T7 Strain Retains Virulence in Macrophages and Mice
To investigate whether chromosomal integration of T7 RNA polymerase affects Salmonella virulence, we first performed macrophage survival assays. J774A.1 macrophages were infected with Salmonella-T7, wild-type 14028s, or the ΔmgtC mutant, which has lost pathogenic activity [16], and intracellular bacterial replication was quantified. At 22 h post-infection, fold replication (CFU T22/T1) of Salmonella-T7 was comparable to the wild-type, whereas ΔmgtC mutants showed significantly reduced survival (Fig. 4A).
To determine whether Salmonella-T7 impacts intracellular bacterial dynamics, we utilized the pFCcGi plasmid, a reporter system that measures non-replicating bacteria during host infection via a fluorescence dilution assay. This dual-fluorescence reporter constitutively expresses mCherry and arabinose-inducible GFP (Fig. 4B). Following macrophage infection, bacteria were analyzed by flow cytometry to determine replication status based on GFP dilution relative to the constitutive mCherry signal (Fig. 4C). Non-replicating bacteria maintain high GFP/mCherry ratios, while replicating bacteria exhibit decreased GFP signals due to dilution. At 2, 6, 9, and 22 h post-infection, ΔmgtC mutants consistently maintained high GFP/mCherry ratios, indicating non-replication. However, Salmonella-T7 strains displayed progressive GFP dilution over time, indicating active replication similar to the wild-type (Figs. 4D and S1). These findings highlight that introduction of T7 RNA polymerase into Salmonella does not alter bacterial physiology during host infection.
In vivo virulence was assessed by intraperitoneal infection of C3H/HeN mice with 10^3^ CFU of Salmonella-T7, wild-type, or ΔmgtC. Mouse survival was monitored over 21 days post-infection. Salmonella-T7 and wild-type strains exhibited similar lethality profiles, with no significant differences in survival rates (Fig. 4E). Together, these data demonstrate that integration of T7 RNA polymerase does not attenuate Salmonella virulence in vitro or in vivo.
Discussion
In this study, we developed and characterized a novel Salmonella enterica Typhimurium strain (Salmonella-T7) engineered to express T7 RNA polymerase from the chromosome under the control of the lac promoter by using a transposon insertion system [20]. This strain enables IPTG-inducible expression of heterologous proteins from T7 promoter-based vectors, such as the widely used pET series, directly in a pathogenic Salmonella background. Using the pRSF-sfGFP reporter plasmid, we observed strong GFP induction upon IPTG treatment with minimal background expression in its absence, both in rich and minimal media (Fig. 2). The ability to achieve dose-dependent, tunable expression further enhances the utility of Salmonella-T7 for applications requiring precise control over gene expression.
Earlier attempts to integrate T7 RNA polymerase in Salmonella, such as the nirB-regulated system in S. Typhi [22] and the araC-P_BAD_ system in S. Typhimurium [23], provided proof-of-concept for expressing proteins in Salmonella. The broad-host-range approach by Hoang et al. (2007) using mariner transposons [24] also demonstrated the potential for integrating T7 RNAP into diverse bacteria, including Salmonella. Although previous systems using nirB [23] or P_BAD_ [24] promoters demonstrated reporter gene activity through methods such as northern blotting or β-galactosidase assays, they did not confirm heterologous protein expression at the protein level (e.g., by SDS-PAGE or Western blot analysis) or assess virulence in host infection models. In contrast, our system utilizes site-specific Tn7 integration at attTn7, uses the well-characterized plac/lacO promoter for tight IPTG-inducible regulation, and supports seamless expression from widely used pET vectors. Importantly, we show for the first time that chromosomal T7 RNA polymerase expression does not attenuate bacterial virulence in vitro or in vivo (Figs. 4 and S1), positioning Salmonella-T7 as a robust platform for pathogenicity studies and synthetic biology applications. While our in vivo analysis focused on survival outcomes and did not include organ bacterial burden measurements, the consistent lethality profiles support the conclusion that virulence was not notably affected. Future studies incorporating organ CFU quantification may provide additional resolution into tissue-specific infection dynamics.
The development of Salmonella-T7 overcomes several limitations of previous expression systems. Single-vector systems in native hosts often suffer from leaky expression, whereas bipartite systems employing two plasmids require dual antibiotic selection, which could be undesirable for studies of pathogenic bacteria. Our chromosomal integration strategy eliminates the need for plasmid maintenance and additional selection markers, enabling stable and reproducible gene expression studies in pathogenic backgrounds. Furthermore, this system can be applied to study virulence factors and host-pathogen interactions under native physiological conditions. By enabling direct expression of heterologous or engineered proteins in a pathogenic context, Salmonella-T7 provides a unique tool for examining bacterial physiology, secretion systems, and immune evasion strategies. Additionally, it could facilitate the production of antigens or vaccines where maintaining the pathogen’s native virulence and regulatory networks is crucial. As Salmonella serves as a platform for live attenuated vaccine development due to its ability to invade host cells and target cancer cells [25??-28], our findings highlight that this engineered strain, with its precise regulatory control, could contribute to the development of Salmonella as an effective vaccine carrier. Moreover, this approach could be adapted to other non-model Gram-negative pathogens, expanding its utility in microbial pathogenesis research.
In conclusion, we present Salmonella-T7 as a powerful and versatile platform for T7 promoter-driven expression in S. enterica. By enabling robust, IPTG-inducible expression of heterologous proteins directly from widely used pET vectors, this system overcomes key limitations of previous approaches and facilitates the seamless transfer of constructs optimized in E. coli. Its ability to maintain full virulence while supporting tightly regulated gene expression makes it highly suitable for both in vitro and in vivo studies of Salmonella biology. Furthermore, Salmonella-T7 provides a valuable tool for dissecting bacterial physiology and host-pathogen interactions under native conditions, while also offering potential applications in vaccine development and synthetic biology.
Supplemental Materials
Supplementary data for this paper are available on-line only at http://jmb.or.kr.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Dubendorff JW Studier FW 1991 Creation of a T 7 autogene. Cloning and expression of the gene for bacteriophage T 7 RNA polymerase under control of its cognate promoter J. Mol. Biol.219616810.1016/0022-2836(91)90857-32023261 · doi ↗ · pubmed ↗
- 2Studier FW Moffatt BA 1986 Use of bacteriophage T 7 RNA polymerase to direct selective high-level expression of cloned genes J. Mol. Biol.18911313010.1016/0022-2836(86)90385-23537305 · doi ↗ · pubmed ↗
- 3Rosano GL Ceccarelli EA 2014 Recombinant protein expression in Escherichia coli: advances and challenges Front. Microbiol.517210.3389/fmicb.2014.0017224860555 PMC 4029002 · doi ↗ · pubmed ↗
- 4Studier FW 2014 Stable expression clones and auto-induction for protein production in E. coli Methods Mol. Biol.1091173210.1007/978-1-62703-691-7_224203322 · doi ↗ · pubmed ↗
- 5Schlegel S Klepsch M Gialama D Wickstrom D Slotboom DJ de Gier JW 2010 Revolutionizing membrane protein overexpression in bacteria Microb. Biotechnol.340341110.1111/j.1751-7915.2009.00148.x 21255339 PMC 3815807 · doi ↗ · pubmed ↗
- 6Robichon C Luo J Causey TB Benner JS Samuelson JC 2011 Engineering Escherichia coli BL 21(DE 3) derivative strains to minimize E. coli protein contamination after purification by immobilized metal affinity chromatography Appl. Environ. Microbiol.774634464610.1128/AEM.00119-1121602383 PMC 3127686 · doi ↗ · pubmed ↗
- 7Makino T Skretas G Georgiou G 2011 Strain engineering for improved expression of recombinant proteins in bacteria Microb. Cell Fact.103210.1186/1475-2859-10-3221569582 PMC 3120638 · doi ↗ · pubmed ↗
- 8Chamberlin M Mc Grath J Waskell L 1970 New RNA polymerase from Escherichia coli infected with bacteriophage T 7Nature 22822723110.1038/228227 a 04920917 · doi ↗ · pubmed ↗