Identification of Novel β‐Lactam Derivatives as Proteasome Inhibitors for Antitumor Therapy
Yu Cao, Gaoya Xu, Lixin Gao, Jingjing Sun, Lu Zhang, Qiao Tong, Limin Kong, Jiankang Zhang, Yubo Zhou, Li Liao, Liping Fu, Jianjun Xi

TL;DR
Researchers designed and tested new β-lactam-based proteasome inhibitors, finding one compound that effectively stops cancer cell growth by blocking proteasome activity.
Contribution
The paper introduces novel β-lactam derivatives as effective proteasome inhibitors with antitumor potential.
Findings
Compound 66 showed micromolar-range proteasome inhibitory activity.
Compound 66 induced apoptosis in RS4;11 cells by inhibiting proteasome pathways.
Compound 66 had antiproliferative effects with IC50 values below 1 μM against RS4;11 cells.
Abstract
A series of dipeptidic proteasome inhibitors with β‐lactam as the C‐terminus was designed and synthesized. Biochemical evaluations of their effects on chymotrypsin‐like (CT‐L) activity revealed that some of them had inhibitory activity against the proteasome, with IC50 values in the micromolar range. Based on the enzymatic results, structure–activity relationships (SAR) were discussed in detail. Some potent compounds were further selected for antiproliferative assays toward multiple cancer cell lines, with compounds 66 and 78 demonstrating activity against RS4;11 cells and IC50 values of less than 1 μM. Additionally, cellular mechanistic studies indicated that these effects were linked to their ability to inhibit proteasome signal pathways and to induce apoptosis in RS4;11 cells, as demonstrated by flow cytometry. Collectively, these results illustrate that compound 66 is a potent…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
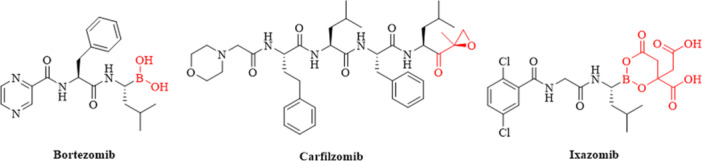 Figure 1
Figure 1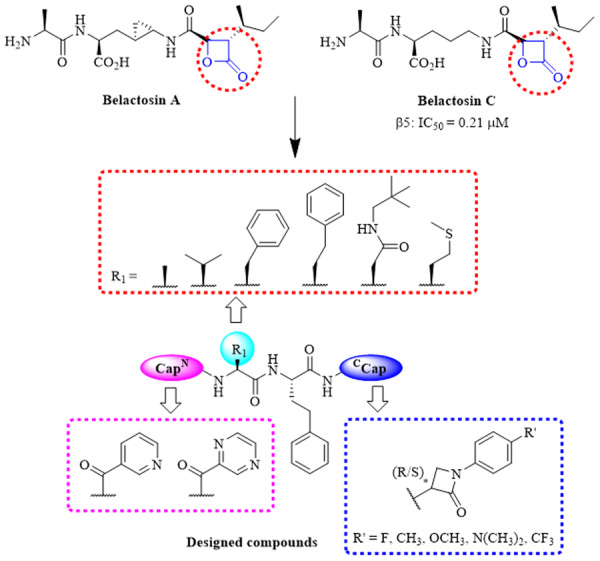 Figure 2
Figure 2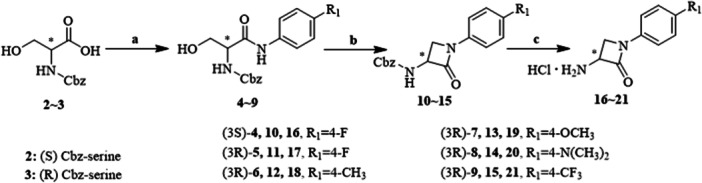 Scheme 1
Scheme 1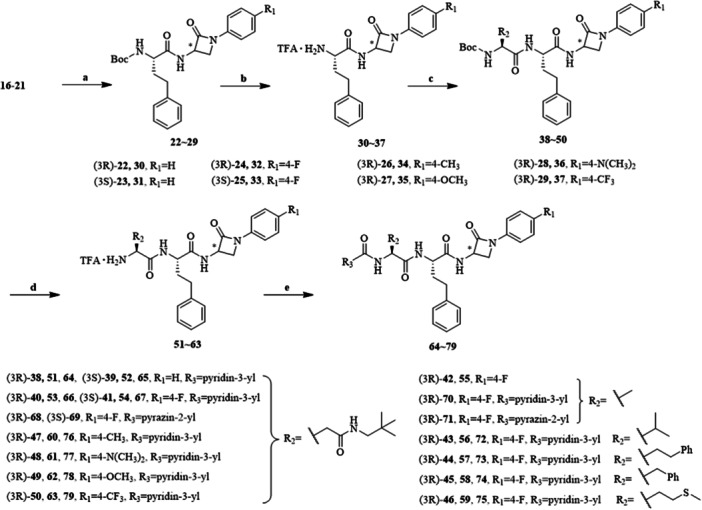 Scheme 2
Scheme 2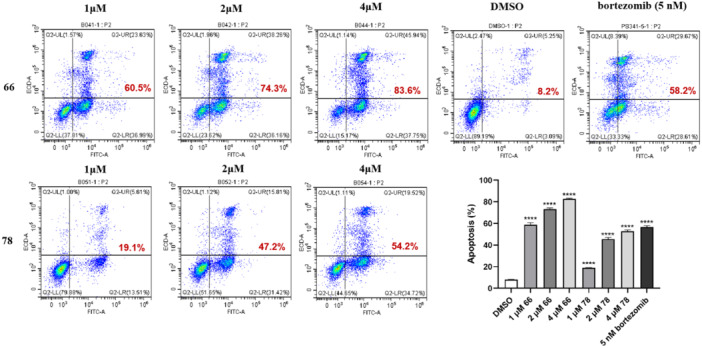 Figure 3
Figure 3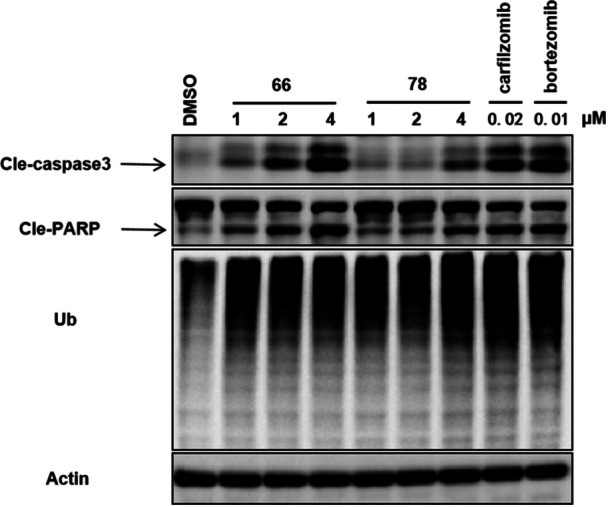 Figure 4
Figure 4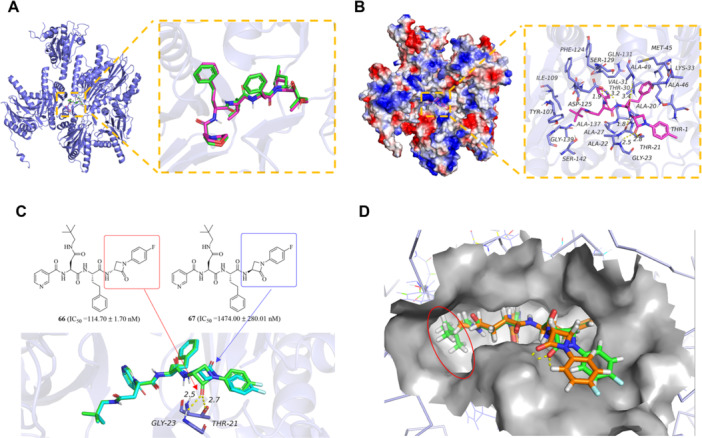 Figure 5
Figure 5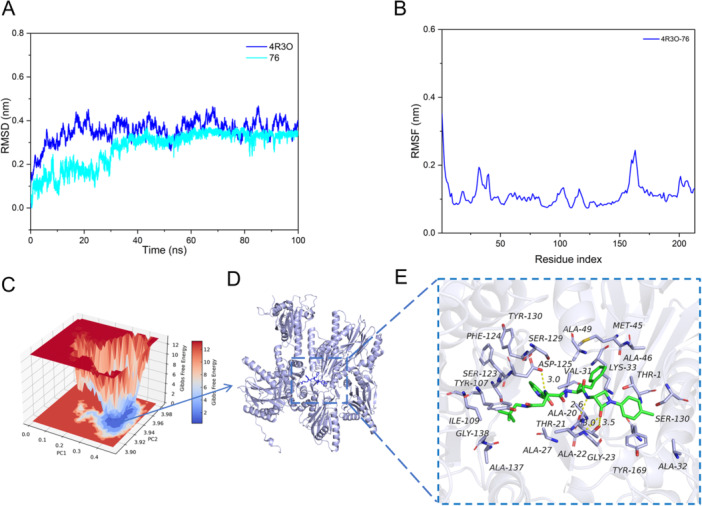 Figure 6
Figure 6|
| ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 |
|
|
|
|
| H | A | Pyridine‐3‐yl | 186.85 ± 35.28 | > 10,000 | > 10,000 |
|
| H | A | Pyridine‐3‐yl | > 10,000 | — | — |
|
| F | A | Pyridine‐3‐yl | 114.70 ± 1.70 | > 10,000 | > 10,000 |
|
| F | A | Pyridine‐3‐yl | 1474.00 ± 280.01 | — | — |
|
| F | A | Pyrazin‐2‐yl | > 10,000 | — | — |
|
| F | A | Pyrazin‐2‐yl | 288.45 ± 57.20 | > 10,000 | > 10,000 |
|
| F | B | Pyridine‐3‐yl | > 10,000 | — | — |
|
| F | B | Pyrazin‐2‐yl | > 10,000 | — | — |
|
| F | C | Pyridine‐3‐yl | > 10,000 | — | — |
|
| F | D | Pyridine‐3‐yl | > 10,000 | — | — |
|
| F | E | Pyridine‐3‐yl | > 10,000 | — | — |
|
| F | F | Pyridine‐3‐yl | > 10,000 | — | — |
|
| CH3 | A | Pyridine‐3‐yl | 72.03 ± 14.89 | > 10,000 | > 10,000 |
|
| N(CH3)2 | A | Pyridine‐3‐yl | 132.25 ± 14.78 | > 10,000 | > 10,000 |
|
| OCH3 | A | Pyridine‐3‐yl | 141.60 ± 30.12 | > 10,000 | > 10,000 |
|
| CF3 | A | Pyridine‐3‐yl | 282.35 ± 33.60 | > 10,000 | > 10,000 |
| Bortezomib | — | — | — | 14.81 ± 0.35 | — | — |
| Carfilzomib | — | — | — | 8.62 ± 5.35 | 338.90 ± 406.00 | 584.00 ± 539.30 |
| IC50 (μM) | |||||
|---|---|---|---|---|---|
| Compound | MV‐4‐11 | RPMI‐8226 | Molt4 | RS4;11 | PBMC‐3Day |
|
| 7.42 ± 0.62 | 5.52 ± 1.12 | 8.34 ± 0.18 | 1.09 ± 0.31 | — |
|
| 1.78 ± 0.19 | 3.02 ± 0.14 | 2.68 ± 0.73 | 0.54 ± 0.18 | 4.90 ± 0.68 |
|
| > 20 | 10.18 ± 0.76 | > 10 | 1.52 ± 0.67 | — |
|
| > 20 | 8.05 ± 1.17 | > 10 | 0.86 ± 0.24 | — |
|
| 2.78 ± 0.41 | 2.34 ± 0.34 | 3.78 ± 0.27 | 0.50 ± 0.13 | 17.18 ± 1.93 |
|
| 2.75 ± 0.46 | 3.44 ± 0.44 | 2.37 ± 0.23 | 0.99 ± 0.17 | — |
| Bortezomib | 0.00515 ± 0.00109 | 0.00364 ± 0.00052 | 0.01327 ± 0.00247 | 0.00587 ± 0.00093 | 0.00157 ± 0.00019 |
- —This study was funded by the Construction Fund of Key Medical Disciplines of Hangzhou (2025HZZD13), the Zhejiang Provincial Natural Science Foundation of China (LTGY24B020001), the Medical Health Scie
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsUbiquitin and proteasome pathways · HIV/AIDS drug development and treatment · Coenzyme Q10 studies and effects
Introduction
1
The 26S proteasome is a sophisticated multi‐catalytic protease complex that operates in an ATP‐dependent manner, playing a vital role in the ubiquitin‐dependent conversion of cellular proteins [1]. It is composed of a 20S core particle flanked by two 19S regulatory subunits. The two 19S regulatory caps are equipped with numerous ATPase active sites and ubiquitin binding domains, which facilitate the recognition and unfolding of ubiquitinated proteins [2, 3]. The 20S core catalytic complex consists of two outer α rings and two inner β rings, with each ring containing seven α and β‐type subunits, respectively. Each β ring contains three proteolytically active β subunits exhibiting different substrate preferences: chymotrypsin‐like (CT‐L) on the β5 subunit, trypsin‐like (T‐L) on the β2 subunit, and caspase‐like (C‐L) on the β1 subunit [4, 5]. The CT‐L activity serves as the rate‐limiting step in the degradation of intracellular proteins, including those that contribute to tumor growth and survival [6, 7]. Consequently, the active sites of the 26S proteasome have been identified as potential therapeutic targets for a variety of diseases, including cancers [8, 9, 10, 11].
For malignant tumor cells to expand, they rely on a markedly elevated rate of protein turnover. Besides rapid cell proliferation, tumor cells also feature a plethora of chromosomal and molecular abnormalities, which result in the generation of numerous distinct protein isoforms [12, 13, 14]. Collectively, these factors result in dysregulated protein expression, thereby imposing a significant burden on the ubiquitin‐proteasome system (UPS) [15]. Such considerations lay the groundwork for therapeutic strategies that target the UPS, particularly through the use of proteasome inhibitors (PIs) [16, 17]. It has been demonstrated that PIs can surmount resistance to specific chemotherapeutic drugs. PIs bortezomib, carfizomib, and ixazomib have been used in the treatment of hematological malignancy (MM) as the first‐line drug [9, 18, 19, 20] (Figure 1).
PIs in clinics.
Nonetheless, the clinical applications of these compounds have been hindered by resistance and severe side effects, particularly in peripheral nerves, cardiac tissue, and the gastrointestinal tract [21, 22, 23]. The unique pharmacophore boronic acid and epoxyketone could form tetrahedral adducts and six‐membered morpholine ring with the active site of threonines, respectively; however, these interactions may cause serious neurotoxicity and cardiotoxicity [2, 9, 24]. Because of the limitations and challenges of the PIs currently approved for MM treatment, there is a need to discover novel PIs that are more effective and safer.
In 2000, Asai and colleagues isolated belactosin A and C from a Streptomyces species [25, 26], as illustrated in Figure 2. Both of them inhibited the CT‐L activity of rabbit 20S proteasome. The inhibitory potency of belactosins toward the proteasome appears to be attributed to the β‐lactone moiety, which undergoes nucleophilic ring opening by the hydroxyl group of the N‐terminal threonine residue, resulting in the formation of a covalent bond [27]. Considering the instability of the lactones, we attempted to replace the lactone ring with the β‐lactam ring. Despite the significant role of β‐lactam rings in antibacterial agents, their application in other areas of medicinal chemistry has remained surprisingly limited [28]. Herein, we report peptidomimetic inhibitors of proteasome bearing a C‐terminal β‐lactam moiety. We examined the effect of various peptide skelectons on the activity, as well as the influence of the stereochemistry at the β‐lactam C‐3 stereocenter and the electronic effect of the N‐1 aryl substituent (Figure 2). Alongside the evaluation of the activity in biochemical and cellular assays, the inhibition mechanism of the most potent compounds was investigated.
The design of β‐lactam derivatives.
Results and Discussion
2
Chemistry
2.1
β‐Lactams 1621 were synthesized through a three‐step reaction sequence initiated from Cbz‐protected serine. This method permits the preparation of enantiopure (3R)‐ or (3S)‐β‐lactams using the predetermined chirality of (R)‐ or (S)‐serine, respectively. Furthermore, this approach yields β‐lactams featuring an aryl substituent at the N‐1 position and a free amino group at the C‐3 position, which enables coupling to the peptides (Scheme 1). The first step involves the coupling of an aryl amine to the C‐terminus of serine, employing COMU as a coupling reagent and TMP as a base. The critical step is the cyclization of (R)‐ or (S)‐amides 49 into (3R)‐ or (3S)‐β‐lactams 1015. This transformation was facilitated through the addition of 1,1‐sulfonyldiimidazole and sodium hydride. Finally, the Cbz‐protection was removed utilizing hydrogen and palladium on charcoal, resulting in (3R)‐ or (3S)‐3‐amino‐β‐lactams 1621.
Synthesis of β‐lactams 16~21. Reagents and conditions: (a) COMU, TMP, corresponding aniline, DMF, 0°C‐rt; (b) Im2SO2, DMF, 0°C; NaH, −20°C; (c) Pd/C, H2, EtOH, rt.
The targeted compounds 6479 were synthesized as shown in Scheme 2. Initially, β‐lactams 1621 were coupled to Boc‐l‐homophenylalanine using EDCI as a coupling reagent and DIPEA as a base. (3R)‐ or (3S)‐β‐lactam derivatives 3037 were obtained after Boc deprotection using trifluoroacetic acid. Boc‐protected amino acids were coupled to intermediates 3037, followed by cleaving Boc‐protection, yielding (3R)‐ or (3S)‐dipeptide‐β‐lactam derivatives 5163. Dipeptide‐β‐lactam derivatives 5163 reacted with nicotinic acid or 2‐pyrazinecarboxylic acid under the previously described conditions to afford final products 64~79.
Synthesis of target compounds 64~79. Reagents and conditions: (a) HOBt, EDCI, Boc‐l‐homophenylalanine, DIPEA, DCM, 0°C‐rt; (b) TFA, DCM, 0°C‐rt; (c) HOBt, EDCI, corresponding amino acids, DIPEA, DCM, 0°C‐rt; (d) TFA, DCM, 0°C‐rt; (e) HOBt, EDCI, nicotinic acid or 2‐pyrazinecarboxylic acid, DIPEA, DCM, 0°C‐rt.
Pharmacology/Biology
2.2
The Inhibitory Activities Against Proteasome and SAR Analysis
2.2.1
As illustrated in Table 1, the target compounds 6479 were screened for their in vitro proteasome CT‐L inhibitory activities, with bortezomib utilized as a positive control. In the initial series of compounds 6469, which featured enantiopure (3R)‐ and (3S)‐ β‐lactams at the C‐terminal, those substituted with (3S)‐β‐lactams showed markedly diminished activity (IC_50_: > 10,000 nM for 65 vs. IC_50_: 186.85 ± 35.28 nM for 64; IC_50_: 1474.00 ± 280.01 nM for 67 vs. IC_50_: 114.70 ± 1.70 nM for 66; IC_50_: > 10,000 nM for 68 vs. IC_50_: 288.45 ± 57.20 nM for 69). The data underscore the critical influence of steric configurations at the C‐3 position on the retention of activity.
Subsequently, when (3R)‐ β‐lactam was retained at the C‐terminal and the F group was incorporated into the benzene ring, further exploration of the dipeptide skeleton was undertaken in the second round of modifications. Regrettably, among the various substitutions (A–F) introduced at the R_2_ position, only the compound with an A substitution exhibited significant inhibitory activity against the proteasome (IC_50_: 114.70 ± 1.70 nM for 66), while compounds featuring other skeletal replacements (B–F) showed no activity (IC_50_: > 10,000 nM for 70, 72–75). Notably, based on the proteasome inhibitory activities of 66 and 69, it was observed that the introduction of pyridine‐3‐yl or pyrazin‐2‐yl at the R_3_ position had minimal impact on the activity.
Building upon the insights gleaned from the first and second rounds of proteasome inhibition assessments, a third round of structural modifications was conducted using compound 66 as the reference, maintaining the R configuration at the C‐3 position, retaining substitution A at the R_2_ position, and incorporating pyridine‐3‐yl at the N‐terminal, while varying the substitutions on the benzene ring. This included both unsubstituted benzene (compound 64) and substitutions with electron‐donating substituents such as methyl (compound 76), dimethylamino (compound 77), and methoxy (compound 78) groups, as well as electron‐withdrawing substitutions including trifluoromethyl (compound 79). The results indicated that diverse substitutions on the benzene ring had a negligible effect on the activity. Notably, the introduction of a methyl group on the benzene resulted in compound 76, which exhibited robust inhibitory activity against the proteasome with an IC_50_ value of 72.03 ± 14.89 nM. To evaluate the target selectivity profile, selected CT‐L inhibitory compounds were further screened against two additional proteolytically active subunits, T‐L on the β2 subunit and C‐L on the β1 subunit, with no detectable inhibitory activity observed.
Antiproliferative Activities
2.2.2
Then, we conducted cellular anti‐proliferation assays of compounds 64, 66, 69, 76, 78, and 79 in malignant hematological tumor cell lines, specifically MV‐4‐11, RPMI‐8226, Molt4, and RS4;11, as well as normal peripheral blood mononuclear cells (PBMC). The cellular activities of target compounds are summarized in Table 2, with bortezomib serving as the reference standard. Among malignant hematological tumor cells, human acute leukemia cell line RS4;11 was more sensitive to the tested compounds, with IC_50_ values recorded at less than 2 μM. Notably, compounds 66 and 78 showed the highest antiproliferative efficacy, achieving IC_50_ values of 0.54 nM and 0.50 nM, respectively. Furthermore, compounds 66 and 78 displayed lower cytotoxicity against normal PBMC compared with (66, IC_50_: 4900 nM; 78, IC_50_: 17180 nM) the positive control bortezomib (IC_50_: 1.57 nM).
Cellular Mechanistic Studies
2.2.3
To further elucidate the potential of compounds 66 and 78 in inducing apoptosis in the human acute leukemia cell line, we assessed their effects on apoptosis in RS4;11 cells via an apoptosis assay. As illustrated in Figure 3, compounds 66 and 78 triggered cell apoptosis in a dose‐dependent manner. Compound 66 exhibited a more pronounced capacity to facilitate apoptosis compared with 78.
*The effect of 66 and 78 on apoptosis of RS4;11. RS4;11 cells were cultured normally or incubated with medium containing 66 (1, 2, and 4 μM), 78 (1, 2, and 4 μM), and 5 nM bortezomib for 12 h. Bar graphs illustrate cell subpopulations labeled with annexin V‐FITC, representing early and late apoptotic stages as assessed by flow cytometry. Results were mean ± SD for three individual experiments. ***p < 0.0001 compared with DMSO (one‐way ANOVA).
Subsequently, we examined the influence of these compounds on apoptosis‐related proteins using western blot assays in RS4;11 cells. The cell line was treated with three concentrations (1, 2, and 4 μM) of compounds 66 and 78 for a duration of 8 h. As depicted in Figure 4, the activation and cleavage of caspase‐3 and PARP were detectable across all samples, indicating their key role in the induction of apoptosis. Notably, the intensity of these signals increased with higher concentrations, corroborating the findings from flow cytometry. Furthermore, 66 and 78 exhibited a significant accumulation of poly‐ubiquitinated proteins, which aligns with that of the positive controls bortezomib and carfilzomib.
The expression of apoptosis‐related proteins Cle‐caspase and Cle‐PARP, treated with increasing concentrations 1, 2, and 4 μM) of 66 and 78, was examined using western blot analysis. Accumulation of ubiquitinated proteins induced by 66 and 78.
Molecular Docking Study
2.3
Molecular docking plays a vital role in elucidating possible protein–target molecule interactions and identifying binding locations. This computational methodology aims to determine the ideal conformation that minimizes the free energy of the system. Through accurate prediction of intermolecular interactions, molecular modeling significantly facilitates the design and development of new therapeutic agents by prioritizing the most promising candidate structures for subsequent experimental validation and development. This docking study was conducted using Glide (Schrödinger version 2021‐2).
The docking study was conducted using the human constitutive 20S proteasome with the PDB ID: 4R3O. A grid box of 15 Å × 15 Å × 15 Å was defined around the native ligand of the selected protein with coordinates (x,y,z): 177.34, −25.55, and 1.82 to encompass the binding region. To validate the docking parameters, we performed redocking experiments using the protein's native ligand. As shown in Figure 5A, the redocked ligand demonstrated excellent structural alignment with the crystallographic conformation (RMSD < 1.78 Å), confirming the reliability of our docking methodology and parameter settings.
Comprehensive analysis of molecules 66, 67, 75, and 76 and human constitutive 20S proteasome (PDB ID code: 4R3O) interactions. (A) The redocking results for 4R3O with nature ligand. (B) The detailed binding mode between 76 and proteasome (PDB ID code: 4R3O). The backbone of the protein was rendered in tube and colored in slate. Compound is rendered in red. (C) The detailed binding mode of molecules 66 and 67 with the complex. The backbone of the protein was rendered in tube and colored in light blue. Compound 66 is rendered in green. Compound 67 is rendered in cyan. (D) Structural comparison of proteasome β5 site occupancy. Compound 66 (green) with neopentyl‐asparagine optimally filling the specificity pocket. Compound 75 (orange) showing inefficient hydrophobic pocket interactions.
Revealing assorted binding interactions, the most bioactive compound 76 (IC_50_ = 72.03 ± 14.89 nM) demonstrated a docking score of −9.619 kcal/mol with the target protein 4R3O. Structural analysis revealed critical hydrogen‐bonding interactions between molecule 76 and key amino acid residues within the binding pocket. Residue THR Z:21 developed hydrogen bonds (1.8 and 2.8 Å) with NH‐proton and the O‐atom in the lactam ring. Likewise, the O‐atom of the lactam ring unveiled one hydrogen bond at a distance of 2.5 Å with residue GLY Z:23. Further, the residue ASP 1:125 produced a hydrogen bond with the NH‐proton of N‐terminus at a distance of 1.9 Å (Figure 5B). A comparison of the docking simulation studies of compounds 66 (IC_50_ = 114.70 ± 1.70 nM) and 67 (IC_50_ = 1474.00 ± 280.01 nM) underscored the significance of chirality. As depicted in Figure 5C, the carbonyl group of molecule 66, containing (3 R)‐ β‐lactam, participated in hydrogen bonding with THR Z:21 (2.7 Å) and GLY Z:23 (2.5 Å). In contrast, molecule 67, which incorporates the (3S)‐ β‐lactam, failed to form hydrogen bonds in the region due to the altered orientation of its carbonyl group. Additionally, we intentionally included compound 75 (IC_50_ > 10,000 nM) as an inactive control for comparative analysis. The striking potency difference between compound 66 (IC_50_ = 114.70 ± 1.70 nM), featuring a neopentyl‐asparagine group at the R_2_ position, and the inactive methionine‐containing analog (compound 75) can be structurally rationalized by the superior complementarity of the neopentyl‐asparagine moiety within the hydrophobic binding pocket (Figure 5D).
Molecular Dynamics (MD) Simulation
2.4
MD simulations are effective tools to uncover the intricate dynamics and stability of protein–ligand complexes, essential for drug development. In this study, we conducted extensive simulations of the human constitutive 20S proteasome (PDB ID code: 4R3O) and molecule 76 using Gromacs 2020, spanning a simulation duration of 100 ns to capture the dynamic changes in the system over time. In our investigation of 4R3O‐76 complex, the root mean square deviation (RMSD) values for the ligands remained consistently below 4 Å throughout the simulation, indicating structural stability (Figure 6A). To monitor local changes in the protein chains, the root mean square fluctuation (RMSF) parameter was employed. The RMSF analysis further revealed fluctuations, particularly around residue index 170 and within the range of 200‐300, suggesting these dynamic variations stem from the positioning of these amino acids in the protein's hinge region. In contrast, most residues exhibit minimal conformational changes, indicating that molecule 76 forms a stable complex with the proteasome (Figure 6B).
MD simulation plots for complexes “4R3O‐76” (PDB ID code: 4R3O) interactions. (A) Root mean square deviation (RMSD) value of proteasome (blue) and molecule 76 (cyan). (B) Root mean square fluctuation (RMSF) value of the proteasome. (C) The 3D free energy landscape of the complex. (D). The 3D structure of the complex. (E) The detailed binding mode between 76 and proteasome (PDB ID code: 4R3O). The backbone of the protein was rendered in tube and colored in light blue. Compound is rendered in green.
The binding free energy of molecule 76 to the proteasome was calculated to be −30.23 ± 4.54 kcal/mol, with van der Waals interactions contributing predominantly (−58.13 ± 0.46 kcal/mol) (Figure 6C). This indicates that the compound stably occupied the binding pocket, engaging in extensive vdW interactions with surrounding residues. This strong interaction arises primarily due to the compound being deeply inserted into the protein cavity, forming favorable van der Waals contacts with nearby amino acids. Additionally, hydrogen bonding between the compound and the proteasome significantly enhances electrostatic stabilization (−31.17 ± 4.39 kcal/mol). Collectively, these interactions indicate that molecule 76 binds the proteasome with high affinity, forming a stable complex that likely underpins its biological activity (Figure 6D,E).
Conclusion
3
In this study, we aimed to mitigate the severe side effects associated with the approved proteasome inhibitor. Inspired by the β‐lactone moiety of belactosines, we designed and synthesized a series of dipeptides featuring enantiopure (3R)‐ and (3S)‐β‐lactams. Evaluation of the proteasome inhibitory activities revealed that target compounds, which include substitution A and (3R)‐β‐lactam within the peptidomimetic skeleton, exhibited potent proteasome inhibition, with IC_50_ values being below 300 μM. Furthermore, the antitumor assays demonstrated that compounds 66 and 78 exhibited strong inhibitory activity against RS4;11 cells. Fortunately, these compounds displayed minimal potency against PBMC cells, suggesting that they might have no toxic effects. Biochemical analyses confirmed that compounds 66 and 78 could induce apoptosis in RS4;11 cells, as evidenced by flow cytometry, and resulted in massive accumulation of poly‐ubiquitinated proteins. Furthermore, we explored the explicit binding interactions of molecule 66 with proteasome using molecular docking. Our analysis revealed that molecule 66 engages in hydrogen bond interactions with Thr21 and Gly23. These findings indicate that compound 66 holds promise as a potential therapeutic agent for the treatment of cancer through proteasome inhibition. However, a significant limitation of the synthesized compound is its poor stability in rat plasma, indicating rapid enzymatic degradation and chemical hydrolysis. This instability poses a considerable challenge to its in vivo efficacy and pharmacokinetic profile. Future research will focus on addressing this issue through strategic molecular modifications to enhance metabolic stability.
Experimental
4
Chemistry
4.1
General
4.1.1
All reagents and solvents were procured from commercial sources and utilized without any further purification. NMR spectra (see the Supporting Information) were recorded for ^1^H NMR at 400/500 MHz and for ^13^C NMR at 100/125 MHz. For ^1^H NMR, CDCl_3_ (δ = 7.26) and DMSO‐d 6 (δ = 2.50) were employed as internal standards, and data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant in Hz, and integration. HRAM‐MS data were obtained using an Agilent 1290 HPLC‐6224 Time of Flight Mass Spectrometer. High‐performance liquid chromatography (HPLC) analyses were performed on an Agilent 1200 system, which was equipped with a photodiode array detector using a ZORBAX SB‐C18 column (4.6 × 250 mm) and detected at 254 nm wavelength. The samples were eluted with a 70:30 methanol/H_2_O mixture at a flow rate of 1 mL/min. All yields reported are unoptimized and generally represent typical outcomes. The InChI codes of the investigated compounds, together with some biological activity data, are provided as Supporting Information.
General Procedure for the Synthesis of (R)‐ or (S)‐Amides 4~9
4.1.2
Cbz‐protected (S)‐ or (R)‐serine (2 or 3, 1.0 equiv) was dissolved in DMF (4 mL) within the reaction flask. Corresponding anilines (1.0 equiv) and TMP (1.1 equiv) were subsequently introduced to the flask. The temperature was lowered to 0°C, and COMU (1.1 equiv) was then added. The reaction proceeded at 0°C for 1 h, followed by an additional 3 h at room temperature. Ethyl acetate (30 mL) was added, and the resulting solution was washed with 1 M HCl (2 × 25 mL), saturated NaHCO_3_ (2 × 25 mL), and saturated NaCl solution (2 × 25 mL). The organic layer was dried over Na_2_SO_4_ and concentrated by evaporation to yield a solid, which was used directly in the next step without purification.
Benzyl(S)‐{1‐[(4‐fluorophenyl)amino]‐3‐hydroxy‐1‐oxopropan‐2‐yl}carbamate (4): white solid, 65%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 10.08 (s, 1H, NH), 7.65 (d, J = 9.0 Hz, 2H, Ar‐H), 7.37–7.34 (m, 6H, Ar‐H + NH), 7.16–7.13 (m, 2H, Ar‐H), 5.05 (s, 2H, CH_2_), 5.02–5.00 (m, 1H, OH), 4.22 (dd, J = 13.6, 6.4 Hz, 1H, CH), 3.66–3.62 (m, 2H, CH_2_).
Benzyl(R)‐[3‐hydroxy‐1‐oxo‐1‐(p‐tolylamino)propan‐2‐yl]carbamate (6): white solid, 73%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 9.90 (s, 1H, NH), 7.50 (d, J = 8.0 Hz, 2H, Ar‐H), 7.37 (d, J = 4.0 Hz, 3H, Ar‐H), 7.34–7.18 (m, 3H, Ar‐H + NH), 7.10 (d, J = 8.0 Hz, 2H, Ar‐H), 5.04 (s, 2H, CH_2_), 4.97 (t, J = 5.5 Hz, 1H, CH), 4.25–4.19 (m, 1H, CH), 3.69–3.58 (m, 2H, CH_2_), 2.25 (s, 3H, CH_3_).
Benzyl(R)‐{3‐hydroxy‐1‐[(4‐methoxyphenyl)amino]‐1‐oxopropan‐2‐yl}carbamate (7): white solid, 69%,^1^H NMR (400 MHz, DMSO‐d 6) δ 9.85 (s, 1H, NH), 7.52 (d, J = 9.0 Hz, 2H, Ar‐H), 7.41–7.19 (m, 6H, Ar‐H + NH), 6.90–6.85 (m, 2H, Ar‐H), 5.04 (s, 2H, CH_2_), 5.01–4.93 (m, 1H, OH), 4.20 (dd, J = 13.8, 6.0 Hz, 1H, CH), 3.71 (s, 3H, CH_3_), 3.69–3.56 (m, 2H, CH_2_).
Benzyl(R)‐(1‐{[4‐(dimethylamino)phenyl]amino}‐3‐hydroxy‐1‐oxopropan‐2‐yl)carbamatee (8): white solid, 74%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 9.69 (s, 1H, NH), 7.45–7.26 (m, 9H, Ar‐H), 6.68 (d, J = 8.8 Hz, 1H, NH), 5.04 (s, 2H, CH_2_), 4.99–4.93 (m, 1H, OH), 4.19 (dd, J = 13.6, 6.4 Hz, 1H, CH), 3.69–3.57 (m, 2H, CH_2_), 2.84 (s, 6H, CH_3_).
Benzyl(R)‐(3‐hydroxy‐1‐oxo‐1‐{[4‐(trifluoromethyl)phenyl]amino}propan‐2‐yl)carbamate (9): white solid, 70%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 10.41 (s, 1H, NH), 7.84 (d, J = 8.8 Hz, 2H, Ar‐H), 7.69 (d, J = 8.4 Hz, 2H, Ar‐H), 7.44 (d, J = 7.6 Hz, 1H, NH), 7.40–7.20 (m, 5H, Ar‐H), 5.11–5.00 (m, 3H, CH_2_ + OH), 4.26 (dd, J = 13.5, 6.0 Hz, 1H, CH), 3.74–3.62 (m, 2H, CH_2_).
General Procedure for the Synthesis of 3(R)‐ or 3(S)‐β‐Lactams 10~15
4.1.3
To a solution of (R)‐ or (S)‐amides 49 (1.0 equiv) in DMF (3 mL), 1,1′‐sulfonyldiimidazole (1.5 equiv) was added at 0°C. The mixture was stirred for 30 min at 0°C. Then the temperature was lowered to −20°C and NaH (1.5 equiv) was added. After completion of the reaction, the mixture was poured into mixed solvent (MeOH/H_2_O = 100:1). The precipitate was filtered and dried to give a crude solid 1015.
Benzyl (S)‐[1‐(4‐fluorophenyl)‐2‐oxoazetidin‐3‐yl]carbamate (10): white solid, 87%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 8.11 (d, J = 8.4 Hz, 1H, NH), 7.76–7.62 (m, 2H, Ar‐H), 7.43–7.35 (m, 5H, Ar‐H), 7.23 (t, J = 8.4 Hz, 2H, Ar‐H), 5.06 (s, 2H, CH_2_), 4.92–4.84 (m, 1H, CH), 3.94 (t, J = 5.6 Hz, 1H, CH_2_), 3.59 (dd, J = 5.2, 2.8 Hz, 1H, CH_2_).
Benzyl (R)‐[2‐oxo‐1‐(p‐tolyl)azetidin‐3‐yl]carbamate (12): white solid, 87%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 8.10 (d, J = 8.5 Hz, 1H, NH), 7.41–7.29 (m, 5H, Ar‐H), 7.25 (d, J = 8.0 Hz, 2H, Ar‐H), 7.18 (d, J = 8.0 Hz, 2H, Ar‐H), 5.06 (s, 2H, CH_2_), 4.90–4.84 (m, 1H, CH), 3.91 (t, J = 6.0 Hz, 1H, CH_2_), 3.56 (dd, J = 5.5, 3.0 Hz, 1H, CH_2_), 2.27 (s, 3H, CH_3_).
Benzyl (R)‐[1‐(4‐methoxyphenyl)‐2‐oxoazetidin‐3‐yl]carbamate (13): white solid, 54%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 8.10 (d, J = 8.4 Hz, 1H, NH), 7.41–7.28 (m, 7H, Ar‐H), 6.99–6.92 (m, 2H, Ar‐H), 5.06 (s, 2H, CH_2_), 4.89–4.83 (m, 1H, CH), 3.90 (t, J = 5.6 Hz, 1H, CH_2_), 3.73 (s, 3H, CH_3_), 3.55 (dd, J = 5.6, 2.8 Hz, 1H, CH_2_).
Benzyl (R)‐{1‐[4‐(dimethylamino)phenyl]‐2‐oxoazetidin‐3‐yl}carbamate (14): white solid, 70%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 8.09 (d, J = 8.5 Hz, 1H, NH), 7.41–7.34 (m, 5H, Ar‐H), 7.22 (d, J = 9.0 Hz, 2H, Ar‐H), 6.75 (d, J = 9.0 Hz, 2H, Ar‐H), 5.06 (s, 2H, CH_2_), 4.88–4.82 (m, 1H, CH), 3.88 (t, J = 5.5 Hz, 1H, CH_2_), 3.51 (dd, J = 5.0, 2.5 Hz, 1H, CH_2_), 2.87 (s, 6H, CH_3_).
Benzyl (R)‐{2‐oxo‐1‐[4‐(trifluoromethyl)phenyl]azetidin‐3‐yl}carbamate (15): white solid, 70%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 8.19 (d, J = 8.4 Hz, 1H, NH), 7.76 (d, J = 8.4 Hz, 2H, Ar‐H), 7.53 (d, J = 8.4 Hz, 2H, Ar‐H), 7.41–7.34 (m, 5H, Ar‐H), 5.06 (s, 2H, CH_2_), 4.95–4.89 (m, 1H, CH), 4.01 (t, J = 6.0 Hz, 1H, CH_2_), 3.71–3.66 (m, 1H, CH_2_).
General Procedure for the Synthesis of (3R)‐ or (3S)‐3‐Amino‐β‐Lactams 16~21
4.1.4
The 3(R)‐ or 3(S)‐β‐lactams 10~15 were dissolved in ethanol (10 mL), combined with 10% palladium on carbon, and stirred under H_2_ atmosphere for 2‐3 h. The mixture was then filtered, and 1 M HCl (1 mL) was added to the filtrate. The filtrate was evaporated to dryness, yielding a pure product as a hydrochloride salt.
General Procedure for the Synthesis of (3R)‐ or (3S)‐β‐Lactam Derivatives 22~29
4.1.5
A solution of Boc‐l‐homophenylalanine (1.0 equiv) in CH_2_Cl_2_ (20 mL) was treated with HOBt (1.1 equiv) and EDCI (1.65 equiv) at 0°C. The reaction mixture was stirred for 30 min at room temperature. Subsequently, (3R)‐ or (3S)‐3‐amino‐β‐lactams 16~21 (1.0 equiv) and DIPEA (3.0 equiv) were added. After stirring at room temperature for an additional 3 h, the reaction system was washed with aqueous NaHCO_3_ (30 mL) and brine (30 mL). The dichloromethane layer was dried over anhydrous Na_2_SO_4_ and then evaporated in vacuo. The crude product was purified using flash chromatography (petroleum ether/ethyl acetate = 2:1).
tert‐Butyl [(S)‐1‐oxo‐1‐{[(R)‐2‐oxo‐1‐phenylazetidin‐3‐yl]amino}‐4‐phenylbutan‐2‐yl]carbamate (22): white solid, 60%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 8.69 (d, J = 8.2 Hz, 1H, NH), 7.40–7.33 (m, 4H, Ar‐H), 7.28 (t, J = 7.5 Hz, 2H, Ar‐H), 7.19 (dd, J = 7.7, 2.6 Hz, 3H, Ar‐H), 7.13–7.08 (m, 2H, Ar‐H), 5.06–4.99 (m, 1H, CH), 3.97–3.87 (m, 2H, CH + CH_2_), 3.56 (dd, J = 5.3, 2.8 Hz, 1H, CH_2_), 2.71–2.52 (m, 2H, CH_2_), 1.95–1.71 (m, 2H, CH_2_), 1.41 (s, 9H, CH_3_).
tert‐Butyl [(S)‐1‐{[(R)‐1‐(4‐fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]carbamate (24): white solid, 65%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 8.68 (d, J = 8.4 Hz, 1H, NH), 7.42–7.36 (m, 2H, Ar‐H), 7.30–7.15 (m, 7H, Ar‐H), 7.12 (d, J = 8.0 Hz, 1H, NH), 5.05–4.99 (m, 1H, CH), 3.95–3.87 (m, 2H, CH + CH_2_), 3.55 (dd, J = 5.6, 2.8 Hz, 1H, CH_2_), 2.69–2.53 (m, 2H, CH_2_), 1.94–1.73 (m, 2H, CH_2_), 1.41 (s, 9H, CH_3_).
tert‐Butyl [(S)‐1‐{[(S)‐1‐(4‐fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]carbamate (25): white solid, 74%, ^1^H NMR (400 MHz, CDCl_3_) δ 7.35–7.27 (m, 5H, Ar‐H), 7.25–7.17 (m, 3H, Ar‐H + NH), 7.09–7.02 (m, 2H, Ar‐H), 5.19 (d, J = 8.0 Hz, 1H, NH), 5.11–5.04 (m, 1H, CH), 4.12–4.21 (m, 1H, CH), 3.95 (t, J = 5.6 Hz, 1H, CH_2_), 3.60 (dd, J = 5.6, 2.4 Hz, 1H, CH_2_), 2.78–2.66 (m, 2H, CH_2_), 2.24–2.14 (m, 1H, CH_2_), 2.02–1.90 (m, 1H, CH_2_), 1.47 (s, 9H, CH_3_).
tert‐Butyl [(S)‐1‐oxo‐1‐{[(R)‐2‐oxo‐1‐(p‐tolyl)azetidin‐3‐yl]amino}‐4‐phenylbutan‐2‐yl]carbamate (26): white solid, 68%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 8.70 (d, J = 8.4 Hz, 1H, NH), 7.30–7.23 (m, 4H, Ar‐H), 7.21–7.15 (m, 5H, Ar‐H), 7.12 (d, J = 8.0 Hz, 1H, NH), 5.06–4.99 (m, 1H, CH), 3.96–3.86 (m, 2H, CH + CH_2_), 3.52 (dd, J = 5.2, 2.4 Hz, 1H, CH_2_), 2.69–2.53 (m, 2H, CH_2_), 2.27 (s, 3H, CH_3_), 1.96–1.74 (m, 2H, CH_2_), 1.41 (s, 9H, CH_3_).
tert‐Butyl [(S)‐1‐{[(R)‐1‐(4‐methoxyphenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]carbamate (27): white solid, 63%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 8.68 (d, J = 8.4 Hz, 1H, NH), 7.33–7.25 (m, 4H, Ar‐H), 7.21–7.15 (m, 3H, Ar‐H), 7.11 (d, J = 7.6 Hz, 1H, NH), 6.98–6.92 (m, 2H, Ar‐H), 5.05–4.98 (m, 1H, CH), 3.94–3.84 (m, 2H, CH + CH_2_), 3.73 (s, 3H, CH_3_), 3.50 (dd, J = 5.2, 2.8 Hz, 1H, CH_2_), 2.69–2.53 (m, 2H, CH_2_), 1.95–1.73 (m, 2H, CH_2_), 1.41 (s, 9H, CH_3_).
tert‐Butyl [(S)‐1‐{[(R)‐1‐[4‐(dimethylamino)phenyl]‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]carbamate (28): white solid, 70%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 8.68 (d, J = 8.0 Hz, 1H, NH), 7.27 (t, J = 7.5 Hz, 2H, Ar‐H), 7.24–7.15 (m, 5H, Ar‐H), 7.09 (d, J = 7.9 Hz, 1H, NH), 6.74 (d, J = 9.0 Hz, 2H, Ar‐H), 5.03–4.98 (m, 1H, CH), 3.95–3.89 (m, 1H, CH), 3.84 (t, J = 5.5 Hz, 1H, CH_2_), 3.49–3.44 (m, 1H, CH_2_), 2.86 (s, 6H, CH_3_), 2.68–2.52 (m, 2H, CH_2_), 1.94–1.74 (m, 2H, CH_2_), 1.41 (s, 9H, CH_3_).
tert‐Butyl [(S)‐1‐oxo‐1‐{[(R)‐2‐oxo‐1‐[4‐(trifluoromethyl)phenyl]azetidin‐3‐yl]amino}‐4‐phenylbutan‐2‐yl]carbamate (29): white solid, 59%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 8.70 (d, J = 8.0 Hz, 1H, NH), 7.75 (d, J = 8.8 Hz, 2H, Ar‐H), 7.53 (d, J = 8.4 Hz, 2H, Ar‐H), 7.32–7.24 (m, 2H, Ar‐H), 7.22–7.12 (m, 4H, Ar‐H + NH), 5.09–5.01 (m, 1H, CH), 3.98 (t, J = 6.0 Hz, 1H, CH_2_), 3.95–3.88 (m, 1H, CH), 3.67–3.60 (m, 1H, CH_2_), 2.69–2.53 (m, 2H, CH_2_), 1.97–1.75 (m, 2H, CH_2_), 1.41 (s, 9H, CH_3_).
General Procedure for the Synthesis of Deprotected (3R)‐ or (3S)‐β‐Lactam Derivatives 30~37
4.1.6
To a solution of (3R)‐ or (3S)‐β‐lactam derivatives 22~29 in CH_2_Cl_2_ (20 mL), the trifluoroacetic acid (TFA, 10 mL) was added at 0°C. The reaction mixture was then warmed to room temperature and stirred for 1–2 h. The solvent and TFA were evaporated under reduced pressure, and ether (20 mL) was added to precipitate a white solid. The resulting solid was filtered and dried and was subsequently used in the next step without further purification.
General Procedure for the Synthesis of Dipeptides 38~50
4.1.7
Boc‐protected amino acids (1.0 equiv) were dissolved in CH_2_Cl_2_ (20 mL), and HOBt (1.1 equiv), along with EDCI (1.65 equiv), was added at 0°C. The reaction was allowed to proceed for 30 min at room temperature. Subsequently, deprotected (3R)‐ or (3S)‐β‐lactam derivatives 30~37 (1.0 equiv) and DIPEA (3.0 equiv) were added. After stirring at room temperature for an additional 3 h, the reaction mixture was washed with aqueous NaHCO_3_ (30 mL) and brine (30 mL). The dichloromethane layer was dried over anhydrous Na_2_SO_4_ and then evaporated in vacuo. The crude product was purified using flash chromatography (dichloromethane/ethyl acetate = 3:1).
tert‐Butyl [(S)‐4‐(neopentylamino)‐1,4‐dioxo‐1‐{[(S)‐1‐oxo‐1‐{[(R)‐2‐oxo‐1‐phenylazetidin‐3‐yl]amino}‐4‐phenylbutan‐2‐yl]amino}butan‐2‐yl]carbamate (38): white solid, 64%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 8.77 (d, J = 8.4 Hz, 1H, NH), 8.22 (d, J = 8.1 Hz, 1H, NH), 7.85 (t, J = 6.1 Hz, 1H, NH), 7.39–7.33 (m, 4H, Ar‐H), 7.26 (t, J = 7.5 Hz, 2H, Ar‐H), 7.17 (t, J = 5.6 Hz, 3H, Ar‐H), 7.09 (ddd, J = 13.2, 5.9, 4.1 Hz, 2H, Ar‐H + NH), 5.10–5.04 (m, 1H, CH), 4.26 (q, J = 7.3 Hz, 1H, CH), 4.21–4.13 (m, 1H, CH), 3.94 (t, J = 5.7 Hz, 1H, CH_2_), 3.58 (dd, J = 5.5, 2.9 Hz, 1H, CH_2_), 2.82 (dd, J = 13.1, 6.4 Hz, 1H, CH_2_), 2.72 (dd, J = 13.1, 6.0 Hz, 1H, CH_2_), 2.68–2.60 (m, 2H, CH_2_), 2.59–2.52 (m, 2H, CH_2_), 2.12–2.01 (m, 1H, CH_2_), 1.88–1.78 (m, 1H, CH_2_), 1.37 (s, 9H, CH_3_), 0.73 (s, 9H, CH_3_).
tert‐Butyl {(S)‐4‐(neopentylamino)‐1,4‐dioxo‐1‐[((S)‐1‐oxo‐1‐{[(S)‐2‐oxo‐1‐phenylazetidin‐3‐yl]amino}‐4‐phenylbutan‐2‐yl)amino]butan‐2‐yl}carbamate (39): white solid, 67%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 8.80 (d, J = 8.6 Hz, 1H, NH), 8.27 (d, J = 7.8 Hz, 1H, NH), 7.88 (t, J = 6.1 Hz, 1H, NH), 7.38–7.32 (m, 4H, Ar‐H), 7.26 (t, J = 7.5 Hz, 2H, Ar‐H), 7.20–7.14 (m, 3H, Ar‐H), 7.11–7.06 (m, 1H, Ar‐H), 7.01 (d, J = 7.7 Hz, 1H, NH), 5.15–5.07 (m, 1H, CH), 4.25 (dd, J = 14.3, 7.3 Hz, 1H, CH), 4.15–4.07 (m, 1H, CH), 3.93 (t, J = 5.6 Hz, 1H, CH_2_), 3.53 (dd, J = 5.4, 2.8 Hz, 1H, CH_2_), 2.78 (dd, J = 13.1, 6.4 Hz, 1H, CH_2_), 2.73–2.61 (m, 3H, CH_2_), 2.60–2.52 (m, 2H, CH_2_), 2.13–2.01 (m, 1H, CH_2_), 1.90–1.80 (m, 1H, CH_2_), 1.36 (s, 9H, CH_3_), 0.70 (s, 9H, CH_3_).
tert‐Butyl [(S)‐1‐{[(S)‐1‐{[(R)‐1‐(4‐fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]amino}‐4‐(neopentylamino)‐1,4‐dioxobutan‐2‐yl]carbamate (40): white solid, 53%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 8.80 (d, J = 8.8 Hz, 1H, NH), 8.27 (d, J = 7.6 Hz, 1H, NH), 7.88 (t, J = 6.0 Hz, 1H, NH), 7.41–7.34 (m, 2H, Ar‐H), 7.30–7.21 (m, 4H, Ar‐H), 7.20–7.14 (m, 3H, Ar‐H), 7.00 (d, J = 7.6 Hz, 1H, NH), 5.16–5.08 (m, 1H, CH), 4.25 (dd, J = 14.4, 7.2 Hz, 1H, CH), 4.16–4.09 (m, 1H, CH), 3.92 (t, J = 5.6 Hz, 1H, CH_2_), 3.52 (dd, J = 5.2, 2.8 Hz, 1H, CH_2_), 2.77 (dd, J = 13.2, 6.4 Hz, 1H, CH_2_), 2.73–2.60 (m, 3H, CH_2_), 2.59–2.52 (m, 2H, CH_2_), 2.13–2.03 (m, 1H, CH_2_), 1.93–1.78 (m, 1H, CH_2_), 1.36 (s, 9H, CH_3_), 0.70 (s, 9H, CH_3_).
tert‐Butyl [(S)‐1‐{[(S)‐1‐{[(S)‐1‐(4‐fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]amino}‐4‐(neopentylamino)‐1,4‐dioxobutan‐2‐yl]carbamate (41): white solid, 59%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 8.79 (d, J = 8.4 Hz, 1H, NH), 8.24 (d, J = 8.0 Hz, 1H, NH), 7.87 (t, J = 6.0 Hz, 1H, NH), 7.43–7.35 (m, 2H, Ar‐H), 7.32–7.23 (m, 4H, Ar‐H), 7.23–7.15 (m, 3H, Ar‐H), 7.08 (d, J = 8.0 Hz, 1H, NH), 5.14–5.06 (m, 1H, CH), 4.28 (dd, J = 14.8, 7.2 Hz, 1H, CH), 4.23–4.15 (m, 1H, CH), 3.96 (t, J = 5.6 Hz, 1H, CH_2_), 3.60 (dd, J = 5.6, 2.8 Hz, 1H, CH_2_), 2.84 (dd, J = 13.2, 6.4 Hz, 1H, CH_2_), 2.74 (dd, J = 13.2, 6.4 Hz, 1H, CH_2_), 2.71–2.62 (m, 2H, CH_2_), 2.62–2.54 (m, 2H, CH_2_), 2.13–2.04 (m, 1H, CH_2_), 1.94–1.81 (m, 1H, CH_2_), 1.39 (s, 9H, CH_3_), 0.75 (s, 9H, CH_3_).
tert‐Butyl [(S)‐1‐{[(S)‐1‐{[(R)‐1‐(4‐fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]amino}‐1‐oxopropan‐2‐yl]carbamate (42): white solid, 73%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 8.66 (d, J = 8.0 Hz, 1H, NH), 7.99 (d, J = 7.9 Hz, 1H, NH), 7.42–7.35 (m, 2H, Ar‐H), 7.31–7.20 (m, 4H, Ar‐H), 7.20–7.14 (m, 3H, Ar‐H), 7.07 (d, J = 6.8 Hz, 1H, NH), 5.04–4.97 (m, 1H, CH), 4.27–4.19 (m, 1H, CH), 4.05–3.96 (m, 1H, CH), 3.92 (t, J = 5.6 Hz, 1H, CH_2_), 3.56 (dd, J = 5.2, 2.8 Hz, 1H, CH_2_), 2.69–2.52 (m, 2H, CH_2_), 2.01–1.81 (m, 2H, CH_2_), 1.35 (s, 9H, CH_3_), 1.20 (d, J = 7.2 Hz, 3H, CH_3_).
tert‐Butyl [(S)‐1‐{[(S)‐1‐{[(R)‐1‐(4‐fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]amino}‐3‐methyl‐1‐oxobutan‐2‐yl]carbamate (43): white solid, 65%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 8.71 (d, J = 8.0 Hz, 1H, NH), 8.00 (d, J = 8.0 Hz, 1H, NH), 7.42–7.35 (m, 2H, Ar‐H), 7.31–7.21 (m, 4H, Ar‐H), 7.20–7.15 (m, 3H, Ar‐H), 6.85 (d, J = 8.8 Hz, 1H, NH), 5.09–5.00 (m, 1H, CH), 4.33–4.24 (m, 1H, CH), 3.94 (t, J = 5.6 Hz, 1H, CH_2_), 3.82 (t, J = 7.6 Hz, 1H, CH), 3.54 (dd, J = 5.6, 2.8 Hz, 1H, CH_2_), 2.70–2.54 (m, 2H, CH_2_), 2.03–1.80 (m, 2H, CH_2_), 1.38 (s, 9H, CH_3_), 0.87 (d, J = 6.4 Hz, 3H, CH_3_), 0.84 (d, J = 6.8 Hz, 3H, CH_3_).
tert‐Butyl [(S)‐1‐{[(S)‐1‐{[(R)‐1‐(4‐fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]carbamate (44): white solid, 65%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 8.70 (d, J = 8.0 Hz, 1H, NH), 8.06 (d, J = 7.8 Hz, 1H, NH), 7.4–7.33 (m, 2H, Ar‐H), 7.28–7.22 (m, 6H, Ar‐H), 7.21–7.15 (m, 7H, Ar‐H + NH), 5.03–4.95 (m, 1H, CH), 4.25 (dd, J = 12.7, 8.5 Hz, 1H, CH), 3.98 (dd, J = 13.2, 7.9 Hz, 1H, CH), 3.91 (t, J = 5.6 Hz, 1H, CH_2_), 3.54 (dd, J = 5.4, 2.7 Hz, 1H, CH_2_), 2.68–2.53 (m, 4H, CH_2_), 1.98–1.80 (m, 4H, CH_2_), 1.38 (s, 9H, CH_3_).
tert‐Butyl [(S)‐1‐{[(S)‐1‐{[(R)‐1‐(4‐fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]amino}‐1‐oxo‐3‐phenylpropan‐2‐yl]carbamate (45): white solid, 69%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 8.67 (d, J = 8.1 Hz, 1H, NH), 8.14 (d, J = 7.9 Hz, 1H, NH), 7.42–7.37 (m, 2H, Ar‐H), 7.28–7.23 (m, 7H, Ar‐H), 7.22–7.16 (m, 5H, Ar‐H), 7.02 (d, J = 8.4 Hz, 1H, NH), 5.03–4.98 (m, 1H, CH), 4.30–4.20 (m, 2H, CH), 3.93 (t, J = 5.6 Hz, 1H, CH_2_), 3.56 (dd, J = 5.4, 2.7 Hz, 1H, CH_2_), 3.02 (dd, J = 13.8, 4.0 Hz, 1H, CH_2_), 2.77 (dd, J = 13.8, 10.5 Hz, 1H, CH_2_), 2.67–2.55 (m, 2H, CH_2_), 2.00–1.83 (m, 2H, CH_2_), 1.30 (s, 9H, CH_3_).
tert‐Butyl [(S)‐1‐{[(S)‐1‐{[(R)‐1‐(4‐fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]amino}‐4‐(methylthio)‐1‐oxobutan‐2‐yl]carbamate (46): white solid, 63%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 8.63 (d, J = 8.0 Hz, 1H, NH), 8.22 (d, J = 8.0 Hz, 1H, NH), 7.40–7.35 (m, 2H, Ar‐H), 7.30–7.19 (m, 5H, Ar‐H), 7.18–7.14 (m, 3H, Ar‐H), 7.12 (d, J = 7.0 Hz, 1H, NH), 5.06–5.00 (m, 1H, CH), 4.28‐4.21 (m, 1H, CH), 4.07–4.00 (m, 1H, CH), 3.95 (t, J = 5.5 Hz, 1H, CH_2_), 3.54 (dd, J = 5.5, 2.5 Hz, 1H, CH_2_), 2.64–2.56 (m, 1H, CH_2_), 2.55–2.41 (m, 3H, CH_2_), 2.03 (s, 3H, CH_3_), 2.01–1.98 (m, 1H, CH_2_), 1.92–1.78 (m, 3H, CH_2_), 1.33 (s, 9H, CH_3_).
tert‐Butyl [(S)‐4‐(neopentylamino)‐1,4‐dioxo‐1‐{[(S)‐1‐oxo‐1‐{[(R)‐2‐oxo‐1‐(p‐tolyl)azetidin‐3‐yl]amino}‐4‐phenylbutan‐2‐yl]amino}butan‐2‐yl]carbamate (47): white solid, 62%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 8.81 (d, J = 8.5 Hz, 1H, NH), 8.28 (d, J = 8.0 Hz, 1H, NH), 7.89 (t, J = 6.0 Hz, 1H, NH), 7.39–7.33 (m, 4H, Ar‐H), 7.27 (t, J = 7.5 Hz, 2H, Ar‐H), 7.22–7.16 (m, 3H, Ar‐H), 7.12–7.08 (m, 1H, Ar‐H), 7.02 (d, J = 7.5 Hz, 1H, NH), 5.15–5.09 (m, 1H, CH), 4.26 (dd, J = 14.0, 7.0 Hz, 1H, CH), 4.17–4.10 (m, 1H, CH), 3.94 (t, J = 5.5 Hz, 1H, CH_2_), 3.54 (dd, J = 5.0, 2.5 Hz, 1H, CH_2_), 2.79 (dd, J = 13.5, 6.5 Hz, 1H, CH_2_), 2.74–2.62 (m, 3H, CH_2_), 2.61–2.53 (m, 2H, CH_2_), 2.27 (s, 3H, CH_3_), 2.14–2.03 (m, 1H, CH_2_), 1.92–1.82 (m, 1H, CH_2_), 1.37 (s, 9H, CH_3_), 0.71 (s, 9H, CH_3_).
tert‐Butyl [(S)‐1‐{[(S)‐1‐{[(R)‐1‐[4‐(dimethylamino)phenyl]‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]amino}‐4‐(neopentylamino)‐1,4‐dioxobutan‐2‐yl]carbamate (48): white solid, 65%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 8.77 (d, J = 8.4 Hz, 1H, NH), 8.25 (d, J = 8.0 Hz, 1H, NH), 7.86 (t, J = 6.0 Hz, 1H, NH), 7.29–7.23 (m, 2H, Ar‐H), 7.22–7.13 (m, 5H, Ar‐H), 7.01 (d, J = 7.6 Hz, 1H, NH), 6.72 (d, J = 8.8 Hz, 2H, Ar‐H), 5.10–5.04 (m, 1H, CH), 4.25 (dd, J = 14.4, 7.2 Hz, 1H, CH), 4.16–4.07 (m, 1H, CH), 3.85 (t, J = 5.6 Hz, 1H, CH_2_), 3.45 (dd, J = 4.8, 2.4 Hz, 1H, CH_2_), 3.35 (s, 6H, CH_3_), 2.79 (dd, J = 13.2, 6.4 Hz, 1H, CH_2_), 2.74–2.60 (m, 3H, CH_2_), 2.60–2.53 (m, 2H, CH_2_), 2.10–2.01 (m, 1H, CH_2_), 1.93–1.79 (m, 1H, CH_2_), 1.36 (s, 9H, CH_3_), 0.71 (s, 9H, CH_3_).
tert‐Butyl [(S)‐1‐{[(S)‐1‐{[(R)‐1‐(4‐methoxyphenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]amino}‐4‐(neopentylamino)‐1,4‐dioxobutan‐2‐yl]carbamate (49): white solid, 53%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 8.79 (d, J = 8.5 Hz, 1H, NH), 8.26 (d, J = 7.5 Hz, 1H, NH), 7.87 (t, J = 6.0 Hz, 1H, NH), 7.33–7.24 (m, 4H, Ar‐H), 7.22–7.15 (m, 3H), 7.01 (d, J = 7.5 Hz, 1H, NH), 6.94 (d, J = 9.0 Hz, 2H, Ar‐H), 5.13–5.08 (m, 1H, CH), 4.26 (dd, J = 14.0, 7.0 Hz, 1H, CH), 4.17–4.09 (m, 1H, CH), 3.90 (t, J = 5.5 Hz, 1H, CH_2_), 3.73 (s, 3H, CH_3_), 3.50 (dd, J = 5.0, 2.5 Hz, 1H, CH_2_), 2.79 (dd, J = 13.0, 6.5 Hz, 1H, CH_2_), 2.75–2.62 (m, 3H, CH_2_), 2.60–2.53 (m, 2H, CH_2_), 2.11–2.01 (m, 1H, CH_2_), 1.93–1.76 (m, 1H, CH_2_), 1.37 (s, 9H, CH_3_), 0.72 (s, 9H, CH_3_).
tert‐Butyl [(S)‐4‐(neopentylamino)‐1,4‐dioxo‐1‐{[(S)‐1‐oxo‐1‐{[(R)‐2‐oxo‐1‐[4‐(trifluoromethyl)phenyl]azetidin‐3‐yl]amino}‐4‐phenylbutan‐2‐yl]amino}butan‐2‐yl]carbamate (50): white solid, 55%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 8.85 (d, J = 8.8 Hz, 1H, NH), 8.32 (d, J = 7.6 Hz, 1H, NH), 7.91 (t, J = 6.0 Hz, 1H, NH), 7.73 (d, J = 8.8 Hz, 2H, Ar‐H), 7.52 (d, J = 8.8 Hz, 2H, Ar‐H), 7.31–7.21 (m, 5H, Ar‐H), 7.06 (d, J = 8.0 Hz, 1H, NH), 5.21–5.14 (m, 1H, CH), 4.48 (dd, J = 13.6, 7.2 Hz, 1H, CH), 4.13–4.07 (m, 1H, CH), 4.02–3.98 (m, 1H, CH_2_), 3.55–3.47 (m, 1H, CH_2_), 2.73 (dd, J = 12.8, 6.4 Hz, 1H, CH_2_), 2.69–2.61 (m, 3H, CH_2_), 2.60–2.55 (m, 2H, CH_2_), 2.12–2.03 (m, 1H, CH_2_), 1.89–1.82 (m, 1H, CH_2_), 1.37 (s, 9H, CH_3_), 0.66 (s, 9H, CH_3_).
General Procedure for the Synthesis of Deprotected Dipeptides 51~63
4.1.8
To a solution of dipeptides 38~50 in CH_2_Cl_2_ (20 mL), the trifluoroacetic acid (TFA, 10 mL) was added at 0°C. The reaction mixture was then warmed to room temperature and stirred for 1–2 h. The solvent and TFA were evaporated under reduced pressure, and ether (20 mL) was added to precipitate a white solid. The resulting solid was filtered and dried and was subsequently used in the next step without further purification.
General Procedure for the Synthesis of Target Compounds 64~79
4.1.9
Nicotinic acid or 2‐Pyrazinecarboxylic acid (1.0 equiv) was dissolved in CH_2_Cl_2_ (20 mL), and HOBt (1.1 equiv), along with EDCI (1.65 equiv), was added at 0°C. The reaction was allowed to proceed for 30 min at room temperature. Subsequently, deprotected dipeptides 51~63 (1.0 equiv) and DIPEA (3.0 equiv) were added. After stirring at room temperature for an additional 3 h, the reaction mixture was washed with aqueous NaHCO_3_ (30 mL) and brine (30 mL). The dichloromethane layer was dried over anhydrous Na_2_SO_4_ and then evaporated in vacuo. The crude product was purified using flash chromatography (dichloromethane/ethyl acetate = 1:1).
(S)‐N ^4^‐Neopentyl‐2‐(nicotinamido)‐N ^1^‐[(S)‐1‐oxo‐1‐{[(R)‐2‐oxo‐1‐phenylazetidin‐3‐yl]amino}‐4‐phenylbutan‐2‐yl]succinimide (64): white solid, 64%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 9.03 (d, J = 1.6 Hz, 1H, pyridine‐H), 8.96 (d, J = 7.6 Hz, 1H, NH), 8.78 (d, J = 8.4 Hz, 1H, NH), 8.71 (dd, J = 4.8, 1.6 Hz, 1H, pyridine‐H), 8.43 (d, J = 8.0 Hz, 1H, NH), 8.24–8.19 (m, 1H, pyridine‐H), 7.93 (t, J = 6.2 Hz, 1H, NH), 7.51 (dd, J = 7.9, 4.8 Hz, 1H, pyridine‐H), 7.40–7.32 (m, 4H, Ar‐H), 7.26–7.21 (m, 2H, Ar‐H), 7.19–7.13 (m, 3H, Ar‐H), 7.12–7.08 (m, 1H, Ar‐H), 5.12–5.07 (m, 1H, CH), 4.83 (q, J = 7.3 Hz, 1H, CH), 4.25–4.15 (m, 1H, CH), 3.95 (t, J = 5.7 Hz, 1H, CH_2_), 3.60 (dd, J = 5.5, 2.9 Hz, 1H, CH_2_), 2.89–2.80 (m, 2H, CH_2_), 2.78–2.70 (m, 2H, CH_2_), 2.69–2.62 (m, 1H, CH_2_), 2.59–2.52 (m, 1H, CH_2_), 2.13–2.03 (m, 1H, CH_2_), 1.92–1.82 (m, 1H, CH_2_), 0.73 (s, 9H, CH_3_). ^13^C NMR (125 MHz, DMSO‐d 6) δ 172.22, 171.56, 170.28, 165.38, 164.97, 152.52, 149.07, 141.65, 138.59, 135.67, 129.95, 129.68, 128.90, 128.72, 126.31, 124.13, 123.87, 116.63, 55.11, 52.68, 51.35, 50.13, 46.81, 37.64, 33.44, 32.19, 31.70, 27.52. HPLC purity: 100.0%. HRAM‐MS (ESI) m/z: [M+H]^+^ calcd for C_34_H_41_N_6_O_5_ 613.3138; found 613.3127.
(S)‐N ^4^‐Neopentyl‐2‐(nicotinamido)‐N ^1^‐[(S)‐1‐oxo‐1‐{[(S)‐2‐oxo‐1‐phenylazetidin‐3‐yl]amino}‐4‐phenylbutan‐2‐yl]succinimide (65): white solid, 59%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 9.02 (d, J = 1.6 Hz, 1H, pyridine‐H), 8.90 (d, J = 7.6 Hz, 1H, NH), 8.80 (d, J = 8.6 Hz, 1H, NH), 8.71 (dd, J = 4.8, 1.6 Hz, 1H, pyridine‐H), 8.47 (d, J = 7.8 Hz, 1H, NH), 8.24–8.14 (m, 1H, pyridine‐H), 7.92 (t, J = 6.2 Hz, 1H, NH), 7.55–7.47 (m, 1H, pyridine‐H), 7.39–7.34 (m, 4H, Ar‐H), 7.26–7.20 (m, 2H, Ar‐H), 7.16 (dd, J = 6.8, 4.3 Hz, 3H, Ar‐H), 7.10 (ddd, J = 8.5, 6.4, 2.2 Hz, 1H, Ar‐H), 5.12 (ddd, J = 8.6, 5.8, 2.8 Hz, 1H, CH), 4.82 (q, J = 7.2 Hz, 1H, CH), 4.16 (ddd, J = 11.6, 7.9, 4.0 Hz, 1H, CH), 3.95 (t, J = 5.6 Hz, 1H, CH_2_), 3.56 (dd, J = 5.5, 2.8 Hz, 1H, CH_2_), 2.84 (ddd, J = 10.9, 6.7, 3.2 Hz, 2H, CH_2_), 2.76–2.62 (m, 3H, CH_2_), 2.56 (dt, J = 11.3, 7.2 Hz, 1H, CH_2_), 2.13–1.83 (m, 2H, CH_2_), 0.70 (s, 9H, CH_3_). ^13^C NMR (125 MHz, DMSO‐d 6) δ 172.16, 171.47, 170.30, 165.41, 164.95, 152.51, 149.09, 141.72, 138.59, 135.67, 129.95, 129.68, 128.88, 128.72, 126.30, 124.11, 123.87, 116.61, 55.25, 52.52, 51.39, 50.16, 46.54, 37.62, 33.50, 32.24, 31.69, 27.57. HPLC purity: 100.0%. HRAM‐MS (ESI) m/z: [M+H]^+^ calcd for C_34_H_41_N_6_O_5_ 613.3138; found 613.3125.
(S)‐N ^1^‐[(S)‐1‐{[(R)‐1‐(4‐Fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]‐N ^4^‐neopentyl‐2‐(nicotinamido)succinimide (66): white solid, 75%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 9.02 (d, J = 1.5 Hz, 1H, pyridine‐H), 8.90 (d, J = 7.5 Hz, 1H, NH), 8.79 (d, J = 8.5 Hz, 1H, NH), 8.71 (dd, J = 4.5, 1.5 Hz, 1H, pyridine‐H), 8.46 (d, J = 7.5 Hz, 1H, NH), 8.23–8.15 (m, 1H, pyridine‐H), 7.92 (t, J = 6.5 Hz, 1H, NH), 7.53–7.47 (m, 1H, pyridine‐H), 7.39–7.33 (m, 4H, Ar‐H), 7.26–7.20 (m, 2H, Ar‐H), 7.18–7.13 (m, 3H, Ar‐H), 7.12–7.07 (m, 1H, Ar‐H), 5.14–5.10 (m, 1H, CH), 4.82 (q, J = 7.0 Hz, 1H, CH), 4.19–4.12 (m, 1H, CH), 3.94 (t, J = 6.0 Hz, 1H, CH_2_), 3.55 (dd, J = 5.5, 3.0 Hz, 1H, CH_2_), 2.87–2.81 (m, 2H, CH_2_), 2.75–2.69 (m, 2H, CH_2_), 2.69–2.64 (m, 1H, CH_2_), 2.60–2.54 (m, 1H, CH_2_), 2.13–2.03 (m, 1H, CH_2_), 1.93–1.83 (m, 1H, CH_2_), 0.70 (s, 9H, CH_3_). ^13^C NMR (125 MHz, DMSO‐d 6) δ 172.22, 171.56, 170.28, 165.38, 164.97, 152.52, 149.07, 141.65, 138.59, 135.67, 129.95, 129.68, 128.90, 128.72, 126.31, 124.13, 123.87, 116.63, 55.11, 52.68, 51.35, 50.13, 46.81, 37.64, 33.44, 32.19, 31.70, 27.52. HPLC purity: 100.0%. HRAM‐MS (ESI) m/z: [M+H]^+^ calcd for C_34_H_40_N_6_O_5_ 631.3044; found 631.3040.
(S)‐N ^1^‐[(S)‐1‐{[(S)‐1‐(4‐Fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]‐N ^4^‐neopentyl‐2‐(nicotinamido)succinimide (67): white solid, 78%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 9.08–9.02 (m, 2H, pyridine‐H + NH), 8.84 (d, J = 8.5 Hz, 1H, NH), 8.71 (dd, J = 5.0, 1.5 Hz, 1H, pyridine‐H), 8.45 (d, J = 8.0 Hz, 1H, NH), 8.28–8.23 (m, 1H, pyridine‐H), 8.05 (t, J = 6.5 Hz, 1H, NH), 7.50 (dd, J = 7.5, 5.0 Hz, 1H, pyridine‐H), 7.42–7.36 (m, 2H, Ar‐H), 7.26–7.20 (m, 4H, Ar‐H), 7.19–7.13 (m, 3H, Ar‐H), 5.11–5.05 (m, 1H, CH), 4.83 (q, J = 7.5 Hz, 1H, CH), 4.26–4.15 (m, 1H, CH), 3.95 (t, J = 6.0 Hz, 1H, CH_2_), 3.60 (dd, J = 5.0, 2.5 Hz, 1H, CH_2_), 2.90–2.84 (m, 1H, CH_2_), 2.84–2.78 (m, 2H, CH_2_), 2.78–2.73 (m, 1H, CH_2_), 2.69–2.62 (m, 1H, CH_2_), 2.60–2.53 (m, 1H, CH_2_), 2.11–2.02 (m, 1H, CH_2_), 1.93–1.83 (m, 1H, CH_2_), 0.73 (s, 9H, CH_3_). ^13^C NMR (125 MHz, DMSO‐d 6) δ 172.20, 171.46, 170.33, 165.40, 164.76, 157.71, 152.49, 149.15, 141.75, 135.71, 135.22, 129.90, 128.88, 128.71, 126.28, 123.83, 118.26 (d, J = 7.8 Hz), 116.42 (d, J = 22.6 Hz), 55.47, 52.60, 51.58, 50.17, 46.79, 37.67, 33.59, 32.24, 31.70, 27.58. HPLC purity: 100.0%. HRAM‐MS (ESI) m/z: [M+H]^+^ calcd for C_34_H_40_N_6_O_5_ 631.3044; found 631.3042.
(S)‐N ^1^‐[(S)‐1‐{[(R)‐1‐(4‐Fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]‐N ^4^‐neopentyl‐2‐(pyrazine‐2‐carboxamido)succinamide (68): white solid, 80%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 9.20 (d, J = 1.0 Hz, 1H, pyrazine‐H), 8.94–8.87 (m, 3H, pyrazine‐H + NH), 8.76 (dd, J = 2.5, 1.5 Hz, 1H, pyrazine‐H), 8.46 (d, J = 7.5 Hz, 1H, NH), 8.03 (t, J = 6.0 Hz, 1H, NH), 7.41–7.36 (m, 2H, Ar‐H), 7.27–7.19 (m, 4H, Ar‐H), 7.19–7.14 (m, 3H, Ar‐H), 5.18–5.13 (m, 1H, CH), 4.87–4.80 (m, 1H, CH), 4.21–4.15 (m, 1H, CH), 3.95 (t, J = 5.5 Hz, 1H, CH_2_), 3.55 (dd, J = 5.0, 2.5 Hz, 1H, CH_2_), 2.94 (dd, J = 15.0, 6.0 Hz, 1H, CH_2_), 2.85 (dd, J = 14.5, 5.5 Hz, 1H, CH_2_), 2.74–2.66 (m, 2H, CH_2_), 2.65–2.54 (m, 2H, CH_2_), 2.15–2.07 (m, 1H, CH_2_), 1.92–1.82 (m, 1H, CH_2_), 0.60 (s, 9H, CH_3_). ^13^C NMR (125 MHz, DMSO‐d 6) δ 172.04, 170.85, 170.32, 164.77, 162.74, 159.65, 157.74, 148.39, 144.51, 143.92, 143.82, 141.62, 135.22, 128.81 (d, J = 15.3 Hz), 126.33, 118.26 (d, J = 7.9 Hz), 116.42 (d, J = 22.7 Hz), 55.25, 53.03, 50.63, 50.03, 47.24, 37.80, 33.27, 32.11, 31.83, 27.38. HPLC purity: 100.0%. HRAM‐MS (ESI) m/z: [M+H]^+^ calcd for C_33_H_39_FN_7_O_5_ 632.2997; found 632.3014.
(S)‐N ^1^‐[(S)‐1‐{[(S)‐1‐(4‐Fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]‐N ^4^‐neopentyl‐2‐(pyrazine‐2‐carboxamido)succinamide (69): white solid, 80%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 9.23 (s, 1H, pyrazine‐H), 8.98 (d, J = 7.6 Hz, 1H, NH), 8.93 (d, J = 7.2 Hz, 1H, pyrazine‐H), 8.85 (d, J = 8.0 Hz, 1H, NH), 8.79 (d, J = 8.0 Hz, 1H, pyrazine‐H), 8.44 (d, J = 7.6 Hz, 1H, NH), 8.02 (t, J = 5.2 Hz, 1H, NH), 7.40–7.37 (m, 2H, Ar‐H), 7.32–7.21 (m, 4H, Ar‐H), 7.19–7.15 (m, 3H, Ar‐H), 5.15–5.06 (m, 1H, CH), 4.92–4.82 (m, 1H, CH), 4.30–4.19 (m, 1H, CH), 3.96 (t, J = 4.8 Hz, 1H, CH_2_), 3.65–3.54 (m, 1H, CH_2_), 2.93 (dd, J = 14.4, 5.6 Hz, 1H, CH_2_), 2.87–2.74 (m, 3H, CH_2_), 2.72–2.56 (m, 2H, CH_2_), 2.18–2.05 (m, 1H, CH_2_), 1.96–1.81 (m, 1H, CH_2_), 0.69 (s, 9H, CH_3_). ^13^C NMR (100 MHz, DMSO‐d 6) δ 171.50, 170.31, 169.83, 164.20, 162.37, 159.38, 147.85, 144.11, 143.41, 143.35, 141.21, 134.73, 134.71, 128.30 (d, J = 10.4 Hz), 125.82, 117.75 (d, J = 8.0 Hz), 115.92 (d, J = 22.7 Hz), 55.01, 52.28, 50.23, 49.63, 46.31, 37.28, 32.93, 31.69, 31.29, 26.98. HPLC purity: 100.0%. HRAM‐MS (ESI) m/z: [M+H]^+^ calcd for C_33_H_39_FN_7_O_5_ 632.2997; found 632.3015.
N‐[(S)‐1‐{[(S)‐1‐{[(R)‐1‐(4‐Fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]amino}‐1‐oxopropan‐2‐yl]nicotinamide (70): white solid, 55%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 9.05 (d, J = 1.6 Hz, 1H, pyridine‐H), 8.85 (d, J = 6.8 Hz, 1H, NH), 8.71–8.68 (m, 1H, pyridine‐H), 8.64 (d, J = 8.0 Hz, 1H, NH), 8.26–8.17 (m, 2H, pyridine‐H + NH), 7.47 (dd, J = 8.0, 4.8 Hz, 1H, pyridine‐H), 7.44–7.34 (m, 2H, Ar‐H), 7.29–7.20 (m, 4H, Ar‐H), 7.20–7.14 (m, 3H, Ar‐H), 5.06–5.01 (m, 1H, CH), 4.56–4.46 (m, 1H, CH), 4.27–4.20 (m, 1H, CH), 3.93 (t, J = 5.6 Hz, 1H, CH_2_), 3.57 (dd, J = 5.6, 2.8 Hz, 1H, CH_2_), 2.70–2.53 (m, 2H, CH_2_), 2.06–1.80 (m, 2H, CH_2_), 1.39 (d, J = 7.2 Hz, 3H, CH_3_). ^13^C NMR (100 MHz, DMSO‐d 6) δ 172.34, 171.81, 165.13, 164.25, 159.39, 157.00, 151.94, 148.67, 141.23, 135.26, 134.69 (d, J = 2.4 Hz), 129.47, 128.31 (d, J = 2.6 Hz), 125.85, 123.32, 117.80 (d, J = 8.0 Hz), 115.96 (d, J = 22.5 Hz)., 55.05, 52.20, 49.36, 46.60, 33.54, 31.30, 17.42. HPLC purity: 100.0%. HRAM‐MS (ESI) m/z: [M+H]^+^ calcd for C_28_H_29_FN_5_O_4_ 518.2204; found 518.2219.
N‐[(S)‐1‐{[(S)‐1‐{[(R)‐1‐(4‐Fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]amino}‐1‐oxopropan‐2‐yl]pyrazine‐2‐carboxamide (71): white solid, 57%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 9.18 (s, 1H, pyrazine‐H), 8.89 (d, J = 2.4 Hz, 1H, pyrazine‐H), 8.83 (d, J = 7.6 Hz, 1H, NH), 8.79–8.68 (m, 2H, pyrazine‐H + NH), 8.37 (d, J = 8.0 Hz, 1H, NH), 7.43–7.36 (m, 2H, Ar‐H), 7.30–7.20 (m, 4H, Ar‐H), 7.20–7.11 (m, 3H, Ar‐H), 5.07–4.99 (m, 1H, CH), 4.65–4.56 (m, 1H, CH), 4.33–4.25 (m, 1H, CH), 3.93 (t, J = 5.2 Hz, 1H, CH_2_), 3.58 (dd, J = 5.2, 2.4 Hz, 1H, CH_2_), 2.68–2.54 (m, 2H, CH_2_), 2.01–1.83 (m, 2H, CH_2_), 1.41 (d, J = 6.8 Hz, 3H, CH_3_). ^13^C NMR (100 MHz, DMSO‐d 6) δ 172.24, 172.11, 164.70, 162.88, 159.90, 157.50, 148.24, 144.77, 143.93, 143.88, 141.68, 135.18, 128.79, 128.76, 126.35, 118.30 (d, J = 8.1 Hz), 116.43 (d, J = 22.5 Hz), 55.62, 52.80, 49.10, 47.13, 34.14, 31.80, 18.91. HPLC purity: 99.7%. HRAM‐MS (ESI) m/z: [M+H]^+^ calcd for C_27_H_28_FN_6_O_4_ 519.2156; found 519.2175.
N‐[(S)‐1‐{[(S)‐1‐{[(R)‐1‐(4‐Fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]amino}‐3‐methyl‐1‐oxobutan‐2‐yl]nicotinamide (72): white solid, 63%, ^1^H NMR (400 MHz, DMSO‐d 6) δ 9.05 (d, J = 1.6 Hz, 1H, pyridine‐H), 8.72 (dd, J = 4.4, 1.6 Hz, 1H, pyridine‐H), 8.70 (d, J = 8.4 Hz, 1H, NH), 8.66 (d, J = 8.4 Hz, 1H, NH), 8.28 (d, J = 8.0 Hz, 1H, NH), 8.23 (d, J = 8.0 Hz, 1H, pyridine‐H), 7.51 (dd, J = 8.0, 4.8 Hz, 1H, pyridine‐H), 7.44–7.36 (m, 2H, Ar‐H), 7.30–7.21 (m, 4H, Ar‐H), 7.21–7.14 (m, 3H, Ar‐H), 5.11–5.05 (m, 1H, CH), 4.37 (t, J = 8.0 Hz, 1H, CH), 4.31–4.23 (m, 1H, CH), 3.95 (t, J = 5.6 Hz, 1H, CH_2_), 3.54 (dd, J = 5.6, 2.8 Hz, 1H, CH_2_), 2.70–2.57 (m, 2H, CH_2_), 2.24–2.14 (m, 1H, CH), 2.03–1.87 (m, 2H, CH_2_), 0.99 (d, J = 6.0 Hz, 6H, CH_3_). ^13^C NMR (100 MHz, DMSO‐d 6) δ 171.57, 170.91, 165.45, 164.22, 159.39, 157.00, 151.88, 148.62, 141.16, 135.32, 134.64 (d, J = 2.4 Hz), 129.80, 128.29, 125.87, 123.34, 117.79 (d, J = 7.9 Hz), 115.95 (d, J = 22.6 Hz), 59.27, 55.04, 52.21, 46.51, 33.64, 31.25, 29.80, 19.36, 18.89. HPLC purity: 99.5%. HRAM‐MS (ESI) m/z: [M+H]^+^ calcd for C_30_H_33_FN_5_O_4_ 546.2517; found 546.2533.
N‐[(S)‐1‐{[(S)‐1‐{[(R)‐1‐(4‐Fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]nicotinamide (73): white solid, 60%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 9.08 (d, J = 1.6 Hz, 1H, pyridine‐H), 8.88 (d, J = 7.4 Hz, 1H, NH), 8.73 (dd, J = 4.7, 1.5 Hz, 1H, NH), 8.69 (d, J = 8.1 Hz, 1H, pyridine‐H), 8.28 (d, J = 7.8 Hz, 1H, NH), 8.26–8.22 (m, 1H, pyridine‐H), 7.51 (dd, J = 7.8, 4.9 Hz, 1H, pyridine‐H), 7.42–7.36 (m, 2H, Ar‐H), 7.29–7.21 (m, 8H, Ar‐H), 7.20–7.13 (m, 4H, Ar‐H), 5.06–4.99 (m, 1H, CH), 4.57–4.49 (m, 1H, CH), 4.32–4.22 (m, 1H, CH), 3.92 (t, J = 5.6 Hz, 1H, CH_2_), 3.56 (dd, J = 5.4, 2.7 Hz, 1H, CH_2_), 2.79–2.63 (m, 3H, CH_2_), 2.61–2.54 (m, 1H, CH_2_), 2.17–1.94 (m, 3H, CH_2_), 1.94–1.82 (m, 1H, CH_2_). ^13^C NMR (125 MHz, DMSO‐d 6) δ 172.30, 172.08, 165.93, 164.70, 159.63, 157.72, 152.47, 149.20, 141.96, 141.68, 135.79, 135.18, 130.09, 128.79, 126.35, 126.30, 123.84, 118.29, 118.23, 116.53, 116.35, 55.60, 54.03, 52.75, 47.08, 33.96, 33.67, 32.31, 31.80. HPLC purity: 100.0%. HRAM‐MS (ESI) m/z: [M+H]^+^ calcd for C_35_H_35_FN_5_O_4_ 608.2673; found 608.2667.
N‐[(S)‐1‐{[(S)‐1‐{[(R)‐1‐(4‐Fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]amino}‐1‐oxo‐3‐phenylpropan‐2‐yl]nicotinamide (74): white solid, 65%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 8.93 (d, J = 1.5 Hz, 1H, pyridine‐H), 8.91 (d, J = 8.3 Hz, 1H, NH), 8.72 (d, J = 8.1 Hz, 1H, NH), 8.68 (dd, J = 4.8, 1.6 Hz, 1H, pyridine‐H), 8.43 (d, J = 7.8 Hz, 1H, NH), 8.14–8.09 (m, 1H, pyridine‐H), 7.49–7.45 (m, 1H, pyridine‐H), 7.42 – 7.38 (m, 4H, Ar‐H), 7.29–7.24 (m, 6H, Ar‐H), 7.20–7.16 (m, 4H, Ar‐H), 5.08–5.01 (m, 1H, CH), 4.84–4.78 (m, 1H, CH), 4.32–4.25 (m, 1H, CH), 3.94 (t, J = 5.6 Hz, 1H, CH_2_), 3.58 (dd, J = 5.5, 2.8 Hz, 1H, CH_2_), 3.21 (dd, J = 13.8, 3.9 Hz, 1H, CH_2_), 3.01 (dd, J = 13.8, 11.0 Hz, 1H, CH_2_), 2.71–2.55 (m, 2H, CH_2_), 2.06–1.85 (m, 2H, CH_2_). ^13^C NMR (125 MHz, DMSO‐d 6) δ 172.24, 171.81, 165.57, 164.75, 159.63, 157.72, 152.44, 148.99, 141.67, 138.77, 135.59, 135.20, 130.01, 129.67, 128.80, 128.56, 126.75, 126.36, 123.85, 118.30, 118.24, 116.55, 116.37, 55.60, 55.36, 52.87, 47.05, 37.28, 34.15, 31.74. HPLC purity: 98.2%. HRAM‐MS (ESI) m/z: [M+H]^+^ calcd for C_34_H_33_FN_5_O_4_ 594.2517; found 594.2517.
N‐[(S)‐1‐{[(S)‐1‐{[(R)‐1‐(4‐Fluorophenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]amino}‐4‐(methylthio)‐1‐oxobutan‐2‐yl]nicotinamide (75): white solid, 74%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 9.04 (dd, J = 2.0, 0.5 Hz, 1H, pyridine‐H), 8.89 (d, J = 7.0 Hz, 1H, NH), 8.69 (dd, J = 5.0, 1.5 Hz, 1H, pyridine‐H), 8.65 (d, J = 8.0 Hz, 1H, NH), 8.58 (d, J = 8.0 Hz, 1H, NH), 8.18–8.12 (m, 1H, pyridine‐H), 7.40–7.36 (m, 3H, pyridine‐H + Ar‐H), 7.30–7.21 (m, 4H, Ar‐H), 7.20–7.15 (m, 3H, Ar‐H), 5.12–5.07 (m, 1H, CH), 4.54 (q, J = 7.0 Hz, 1H, CH), 4.28–4.20 (m, 1H, CH), 3.94 (t, J = 6.0 Hz, 1H, CH_2_), 3.50 (dd, J = 5.5, 2.5 Hz, 1H, CH_2_), 2.69–2.58 (m, 2H, CH_2_), 2.58–2.51 (m, 2H, CH_2_), 2.07 (s, 3H, CH_3_), 2.06–2.00 (m, 3H, CH_3_), 1.91–1.82 (m, 1H, CH_2_). ^13^C NMR (125 MHz, DMSO‐d 6) δ 172.18, 172.00, 166.03, 164.61, 159.64, 157.73, 152.57, 149.15, 141.55, 135.74, 135.16, 129.64, 128.83, 128.77, 126.41, 123.74, 118.27 (d, J = 8.0 Hz), 116.47 (d, J = 22.6 Hz), 55.52, 54.03, 52.61, 47.30, 33.74, 31.90, 31.47, 30.34, 15.11. HPLC purity: 96.7%. HRAM‐MS (ESI) m/z: [M+H]^+^ calcd for C_30_H_33_FN_5_O_4_S 578.2237; found 578.2245.
(S)‐N ^4^‐Neopentyl‐2‐(nicotinamido)‐N ^1^‐[(S)‐1‐oxo‐1‐{[(R)‐2‐oxo‐1‐(p‐tolyl)azetidin‐3‐yl]amino}‐4‐phenylbutan‐2‐yl]succinamide (76): white solid, 56%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 9.02 (d, J = 1.5 Hz, 1H, pyridine‐H), 8.91 (d, J = 8.0 Hz, 1H, NH), 8.78 (d, J = 8.5 Hz, 1H, NH), 8.71 (dd, J = 5.0, 1.5 Hz, 1H, pyridine‐H), 8.46 (d, J = 8.0 Hz, 1H, NH), 8.22–8.16 (m, 1H, pyridine‐H), 7.91 (t, J = 6.0 Hz, 1H, NH), 7.50 (dd, J = 7.5, 4.5 Hz, 1H, pyridine‐H), 7.26–7.20 (m, 4H, Ar‐H), 7.19–7.10 (m, 5H, Ar‐H), 5.13–5.07 (m, 1H, CH), 4.82 (q, J = 7.0 Hz, 1H, CH), 4.19–4.13 (m, 1H, CH), 3.91 (t, J = 5.5 Hz, 1H, CH_2_), 3.52 (dd, J = 5.5, 3.0 Hz, 1H, CH_2_), 2.87–2.80 (m, 2H, CH_2_), 2.76–2.69 (m, 2H, CH_2_), 2.69–2.63 (m, 1H, CH_2_), 2.60–2.53 (m, 1H, CH_2_), 2.26 (s, 3H, CH_3_), 2.12–2.03 (m, 1H, CH_2_), 1.92–1.83 (m, 1H, CH_2_), 0.71 (s, 9H, CH_3_). ^13^C NMR (125 MHz, DMSO‐d 6) δ 172.19, 171.56, 170.26, 165.38, 164.63, 152.52, 149.08, 141.65, 136.27, 135.67, 133.17, 130.02, 129.94, 128.90, 128.72, 126.30, 123.87, 116.58, 55.08, 52.69, 51.36, 50.13, 46.81, 37.64, 33.47, 32.20, 31.71, 27.52, 20.95. HPLC purity: 99.4%. HRAM‐MS (ESI) m/z: [M+H]^+^ calcd for C_35_H_43_N_6_O_5_ 627.3295; found 627.3303.
(S)‐N ^1^‐[(S)‐1‐{[(R)‐1‐[4‐(Dimethylamino)phenyl]‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]‐N ^4^‐neopentyl‐2‐(nicotinamido)succinamide (77): white solid, 53%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 9.03 (d, J = 1.5 Hz, 1H, pyridine‐H), 8.93 (d, J = 8.0 Hz, 1H, NH), 8.77 (d, J = 8.5 Hz, 1H, NH), 8.73 (dd, J = 4.5, 1.0 Hz, 1H, pyridine‐H), 8.45 (d, J = 7.5 Hz, 1H, NH), 8.26–8.20 (m, 1H, pyridine‐H), 7.91 (t, J = 6.0 Hz, 1H, NH), 7.54 (dd, J = 8.0, 5.0 Hz, 1H, pyridine‐H), 7.28–7.20 (m, 5H, Ar‐H), 7.19–7.12 (m, 4H, Ar‐H), 5.10–5.06 (m, 1H, CH), 4.82 (dd, J = 14.0, 7.0 Hz, 1H, CH), 4.18–4.13 (m, 1H, CH), 3.88 (t, J = 5.5 Hz, 1H, CH_2_), 3.50 (dd, J = 5.0, 2.5 Hz, 1H, CH_2_), 2.90 (s, 6H, CH_3_), 2.86–2.80 (m, 2H, CH_2_), 2.75–2.66 (m, 3H, CH_2_), 2.58–2.52 (m, 1H, CH_2_), 2.09–2.02 (m, 1H, CH_2_), 1.90–1.82 (m, 1H, CH_2_), 0.71 (s, 9H, CH_3_). ^13^C NMR (125 MHz, DMSO‐d 6) δ 172.15, 171.53, 170.23, 165.22, 152.11, 148.71, 141.66, 136.17, 130.10, 128.90, 128.72, 126.31, 124.07, 117.73, 55.05, 52.70, 51.37, 50.12, 46.88, 37.62, 33.50, 32.21, 31.72, 27.54. HPLC purity: 95.8%. HRAM‐MS (ESI) m/z: [M+H]^+^ calcd for C_36_H_46_N_7_O_5_ 656.3560; found 656.3565.
(S)‐N ^1^‐[(S)‐1‐{[(R)‐1‐(4‐Methoxyphenyl)‐2‐oxoazetidin‐3‐yl]amino}‐1‐oxo‐4‐phenylbutan‐2‐yl]‐N ^4^‐neopentyl‐2‐(nicotinamido)succinimide (78): white solid, 56%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 9.03 (d, J = 2.0 Hz, 1H, pyridine‐H), 8.92 (d, J = 7.5 Hz, 1H, NH), 8.78 (d, J = 8.5 Hz, 1H, NH), 8.71 (dd, J = 5.0, 1.5 Hz, 1H, pyridine‐H), 8.47 (d, J = 8.0 Hz, 1H, NH), 8.25–8.16 (m, 1H, pyridine‐H), 7.92 (t, J = 6.0 Hz, 1H, NH), 7.51 (dd, J = 7.5, 5.0 Hz, 1H, pyridine‐H), 7.34–7.28 (m, 2H, Ar‐H), 7.27–7.20 (m, 2H, Ar‐H), 7.19–7.13 (m, 3H, Ar‐H), 6.98–6.91 (m, 2H, Ar‐H), 5.14–5.08 (m, 1H, CH), 4.83 (q, J = 7.0 Hz, 1H, CH), 4.19–4.13 (m, 1H, CH), 3.91 (t, J = 5.5 Hz, 1H, CH_2_), 3.72 (s, 3H, CH_3_), 3.51 (dd, J = 5.5, 2.5 Hz, 1H, CH_2_), 2.89–2.80 (m, 2H, CH_2_), 2.76–2.64 (m, 3H, CH_2_), 2.60–2.52 (m, 1H, CH_2_), 2.14–2.03 (m, 1H, CH_2_), 1.93–1.82 (m, 1H, CH_2_), 0.71 (s, 9H, CH_3_). ^13^C NMR (125 MHz, DMSO‐d 6) δ 172.17, 171.56, 170.25, 165.37, 164.19, 156.02, 152.50, 149.06, 141.65, 135.70, 132.22, 129.94, 128.90, 128.71, 126.30, 123.89, 117.89, 114.90, 55.76, 55.10, 52.69, 51.35, 50.12, 46.96, 37.63, 33.46, 32.21, 31.71, 27.52. HPLC purity: 96.6%. HRAM‐MS (ESI) m/z: [M+H]^+^ calcd for C_35_H_43_N_6_O_6_ 643.3244; found 643.3245.
(S)‐N ^4^‐Neopentyl‐2‐(nicotinamido)‐N ^1^‐[(S)‐1‐oxo‐1‐{[(R)‐2‐oxo‐1‐[4‐(trifluoromethyl)phenyl]azetidin‐3‐yl]amino}‐4‐phenylbutan‐2‐yl]succinamide (79): white solid, 64%, ^1^H NMR (500 MHz, DMSO‐d 6) δ 9.03 (d, J = 2.0 Hz, 1H, pyridine‐H), 8.92 (d, J = 7.5 Hz, 1H, NH), 8.85 (d, J = 8.5 Hz, 1H, NH), 8.72 (dd, J = 5.0, 1.5 Hz, 1H, pyridine‐H), 8.43 (d, J = 8.0 Hz, 1H, NH), 8.22–8.20 (m, 1H, pyridine‐H), 7.95 (t, J = 6.0 Hz, 1H, NH), 7.52–7.50 (m, 1H, pyridine‐H), 7.27–7.20 (m, 4H, Ar‐H), 7.18–7.11 (m, 5H, Ar‐H), 5.22–5.16 (m, 1H, CH), 4.88–4.83 (m, 1H, CH), 4.18–4.12 (m, 1H, CH), 4.03–3.99 (m, 1H, CH_2_), 3.62–3.60 (m, 1H, CH_2_), 2.92 (dd, J = 13.5, 6.5 Hz, 1H, CH_2_), 2.88–2.84 (m, 1H, CH_2_), 2.71–2.68 (m, 2H, CH_2_), 2.61–2.53 (m, 2H, CH_2_), 2.13–2.06 (m, 1H, CH_2_), 1.99–1.94 (m, 1H, CH_2_), 0.68 (s, 9H, CH_3_). ^13^C NMR (125 MHz, DMSO‐d 6) δ 172.12, 171.58, 170.34, 165.35, 152.50, 149.06, 141.61, 135.70, 129.95, 128.91, 128.72, 126.31, 123.92, 116.92, 55.46, 52.54, 51.68, 50.10, 47.11, 37.66, 33.31, 32.12, 31.69, 27.6. HPLC purity: 100.0%. HRAM‐MS (ESI) m/z: [M+H]^+^ calcd for C_35_H_40_F_3_N_6_O_5_ 681.3012; found 681.3029.
Pharmacological/Biological Assays
4.2
In Vitro 20S Proteasome Chymotrypsin‐Like Inhibition Assay
4.2.1
The human constitutive proteasome was generously provided by Dr. Jiang‐ping Wu (Notre‐Dame Hospital, Montreal, QC, Canada). The inhibition of enzyme activity was assessed by monitoring the decrease in hydrolysis of the fluorogenic substrate Suc‐LLVY‐AMC for ChT‐L. All compounds were initially dissolved in DMSO and subsequently diluted with water to achieve a 10% DMSO solution. The proteasome activity assay was performed in a total volume of 50 μL, with all samples diluted in Tris–HCl buffer (100 mM Tris–HCl, pH 8.0). Reactions were conducted in 384‐well polystyrene microplates. 10 μL purified human proteasome (25 μg/mL) was added to 1 μL of each compound; after incubation for 15 min, the mixture was combined with 39 μL of the fluorogenic substrate. The release of the AMC moiety was detected by monitoring the increase of fluorescence with the Envision system, using 355 nm excitation and 460 nm emission wavelengths. The IC_50_ values were calculated using the software GraphPad Prism4, employing the “sigmoidal dose–response (variable slope)” equation for curve fitting.
Cell Proliferation Inhibition Assay
4.2.2
MV‐4‐11 cells (1 × 10^4^ cells/well), RPMI‐8226 cells (8 × 10^3^ cells/well), Molt4 cells (2 × 10^3^ cells/well), RS4;11 cells (2 × 10^4^ cells/well), and PBMC cells (2 × 10^4^ cells/well) were seeded in 96‐well plates (Corning, 3599). Cells were treated with either 0.2% DMSO or fivefold dilutions of compounds from 10 mM stock solutions in DMSO (0.2% final concentration of DMSO) for 72 h. 10 μL of CCK8 reagent was added to each well, and the plates were incubated at 37°C for 3 h. Absorbance was measured at 450 nm and 650 nm using a Spectra Max Molecular Devices microplate reader. The final data were calibrated by OD_450 nm_‐OD_650 nm_. Proliferation inhibition rates were calculated using the following equation: Inhibition ratio = (OD_DMSO_‐OD_Compd_)/(OD_DMSO_‐OD_Blank_) × 100%. The concentrations of the compounds that inhibited cell growth by 50% (IC_50_) were calculated using GraphPad Prism version 9.5.
Apoptosis Assay and Western Blot Analysis
4.2.3
Apoptosis was assessed using an Annexin V‐APC/7AAD apoptosis detection kit (GA1023‐KGA1026, Keygentec, Nanjing, China), followed by flow cytometry analysis. Briefly, cells were treated with the indicated drug concentration and durations or left untreated, then washed twice with PBS, stained with Annexin V‐APC/7AAD, and analyzed using flow cytometry (CytoFLEX flow cytometer, Beckman Coulter Inc.). RS4;11 cells were exposed to various concentrations of the compound or DMSO. Protein lysates were prepared using 4 × Laemmli sample buffer (161073, BIORAD, Richmond, USA). Protein samples were separated by SDS‐polyacrylamide gel electrophoresis (BIO‐RAD, Richmond, USA) and subsequently transferred to nitrocellulose (NC) membranes. Following a 90‐min blocking step with 5% skim milk at room temperature, the NC membranes were incubated with the primary antibody at 4 ^o^C overnight, followed by three washes with TBST for 10 min each. The NC membranes were then incubated with secondary antibodies for 1.5 h at room temperature. Signals were detected by ECL (KF8001&KF8003, Affinity, China) using the BIO‐RAD ChemiDoc Touch Imaging System.
Computational Methods
4.3
Molecular Docking Studies
4.3.1
The molecular docking procedure was conducted using Glide (Schrödinger version 2021‐2) with the default option (standard precision mode). To analyze the binding interaction of target compounds with the active site of proteasome, the protein structure of 20S proteasome was utilized, obtained from the RCSB Protein Data Bank (PDB) as entry 4R3O. The protein was prepared using the Protein Preparation Wizard in Schrödinger's Maestro environment by removing nonrelevant ligands and solvent molecules, retaining crystallographic water molecules involved in the binding site. Missing residues were repaired, bond orders fixed, hydrogen atoms added, and the protein structure optimized with OPLS_2005 force field. For the preparation of ligands, the 3D structures were generated, and their energies were minimized in LigPrep. A grid box measuring 15 Å × 15 Å × 15 Å was created, centered at the native ligand of the selected protein with coordinates (x,y,z): 177.34, −25.55, and 1.82. Interactions of the docked proteasome with the ligand were analyzed, and the docking conformations were selected and saved based on the calculated Glide docking energy score. Graphical images were created using Pymol.
MD Studies
4.3.2
To investigate the interactions within the molecule 76‐proteasome complex, MD simulations were conducted using Gromacs 2020 software. The topology of the proteasome protein was generated using the AMBER99SB‐ILDN force field, while the topology of the ligand was generated by sobtop server using gaff force field. The TIP3P explicit water model was selected, with a minimum distance of 1.0 nm between the protein atoms and the edges of the water box. Sodium or chloride ions were added to neutralize the system charge based on the docking results. Further, energy minimization was done, and the minimized systems were subjected to NPT (Number of particles Pressure Temperature) equilibration of 50 ps. Finally, the systems were subjected to the production MD runs of 100 ns.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
ArchPharm SupplMat InChI.
Supporting_information.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1T. A. Thibaudeau and D. M. Smith , “A Practical Review of Proteasome Pharmacology,” Pharmacological Reviews 71 (2019): 170–197, 10.1124/pr.117.015370.30867233 PMC 6423620 · doi ↗ · pubmed ↗
- 2A. F. Kisselev , W. A. van der Linden , and H. S. Overkleeft , “Proteasome Inhibitors: An Expanding Army Attacking a Unique Target,” Chemistry & Biology 19 (2012): 99–115, 10.1016/j.chembiol.2012.01.003.22284358 PMC 3503453 · doi ↗ · pubmed ↗
- 3R. Kandel , J. Jung , and S. Neal , “Proteotoxic Stress and the Ubiquitin Proteasome System,” Seminars in Cell & Developmental Biology 156 (2024): 107–120, 10.1016/j.semcdb.2023.08.002.37734998 PMC 10807858 · doi ↗ · pubmed ↗
- 4G. Zeng , Q. Yu , R. Zhuang , et al., “Recent Advances and Future Perspectives of Noncompetitive Proteasome Inhibitors,” Bioorganic Chemistry 135 (2023): 106507, 10.1016/j.bioorg.2023.106507.37030106 · doi ↗ · pubmed ↗
- 5H. C. Hsu , J. Wang , A. Kjellgren , H. Li , and G. N. Demartino , “Ηigh‐Resolution Structure of Mammalian PI 31–20S Proteasome Complex Reveals Mechanism of Proteasome Inhibition,” Journal of Biological Chemistry 299 (2023): 104862, 10.1016/j.jbc.2023.104862.37236357 PMC 10319324 · doi ↗ · pubmed ↗
- 6L. J. Crawford , B. Walker , and A. E. Irvine , “Proteasome Inhibitors in Cancer Therapy,” Journal of Cell Communication and Signaling 5 (2011): 101–110, 10.1007/s 12079-011-0121-7.21484190 PMC 3088792 · doi ↗ · pubmed ↗
- 7I. E. Wertz and X. Wang , “From Discovery to Bedside: Targeting the Ubiquitin System,” Cell Chemical Biology 26 (2019): 156–177, 10.1016/j.chembiol.2018.10.022.30554913 · doi ↗ · pubmed ↗
- 8L. Bedford , J. Lowe , L. R. Dick , R. J. Mayer , and J. E. Brownell , “Ubiquitin‐Like Protein Conjugation and the Ubiquitin–Proteasome System as Drug Targets,” Nature Reviews Drug Discovery 10 (2011): 29–46, 10.1038/nrd 3321.21151032 PMC 7097807 · doi ↗ · pubmed ↗