An innovative approach to development of new pyrazolylquinolin-2-one hybrids as dual EGFR and BRAFV600E inhibitors
Mohamed M. Hawwas, Ahmed S. Mancy, Mohamed Ramadan, Tarek S. Ibrahim, Ashraf H. Bayoumi, Mohamed Alswah

TL;DR
This study introduces new compounds that effectively inhibit two cancer-related proteins, showing strong antiproliferative effects in cancer cell lines.
Contribution
The paper presents novel pyrazolylquinolin-2-one hybrids with dual inhibition of EGFR and BRAFV600E, showing high efficacy in cancer cell lines.
Findings
Compound 4e showed the highest antiproliferative activity with IC50 values of 4.982 ± 0.2 to 36.52 ± 1.46 µM in cancer cell lines.
Compound 4e exhibited strong inhibition of EGFR and BRAFV600E with IC50 values of 0.055 ± 0.002 and 0.068 ± 0.003 µM, respectively.
Docking simulations confirmed compound 4e's strong affinity for both EGFR and BRAFV600E with scores of − 3.226 and − 3.474 kcal/mol.
Abstract
Novel quinoline-based derivatives 2a–e and 4a–j have been designed and synthesized as potential antiproliferative agents. The designed compounds were screened for their antiproliferative activity against sixty cell lines according to NCI protocol. The promising hybrids 4d–g are screened by MTT assays on three cancer cell lines: leukemia (MOLT-4), lung cancer (HOP-92), and breast cancer (T47D), with IC50 values ranging from 4.982 ± 0.2 to 36.52 ± 1.46 µM compared to Staurosporine, with compound 4e being the most effective. Derivatives 4d–g were evaluated for their inhibitory activity on EGFR and BRAFV600E. Compound 4e exhibited the highest inhibitory activities, with IC50 values of 0.055 ± 0.002 μM for EGFR and 0.068 ± 0.003 μM for BRAFV600E, compared to the reference drugs erlotinib (IC50 0.06 ± 0.002 μM) and vemurafenib (IC50 0.035 ± 0.001 μM), respectively. Cell cycle analysis of the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
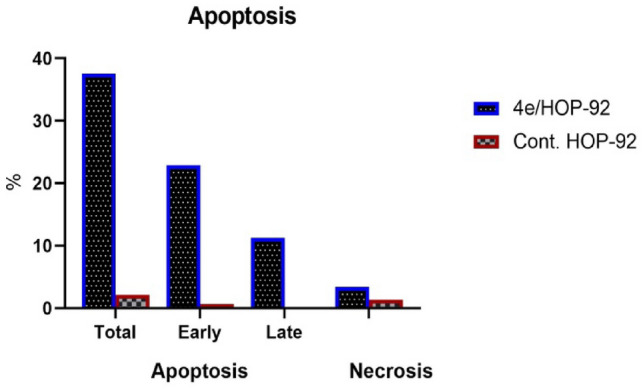 Figure 10
Figure 10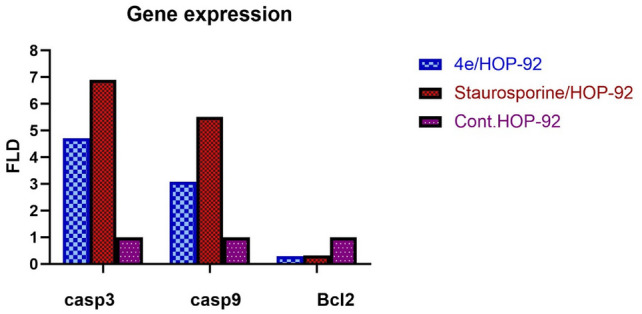 Figure 11
Figure 11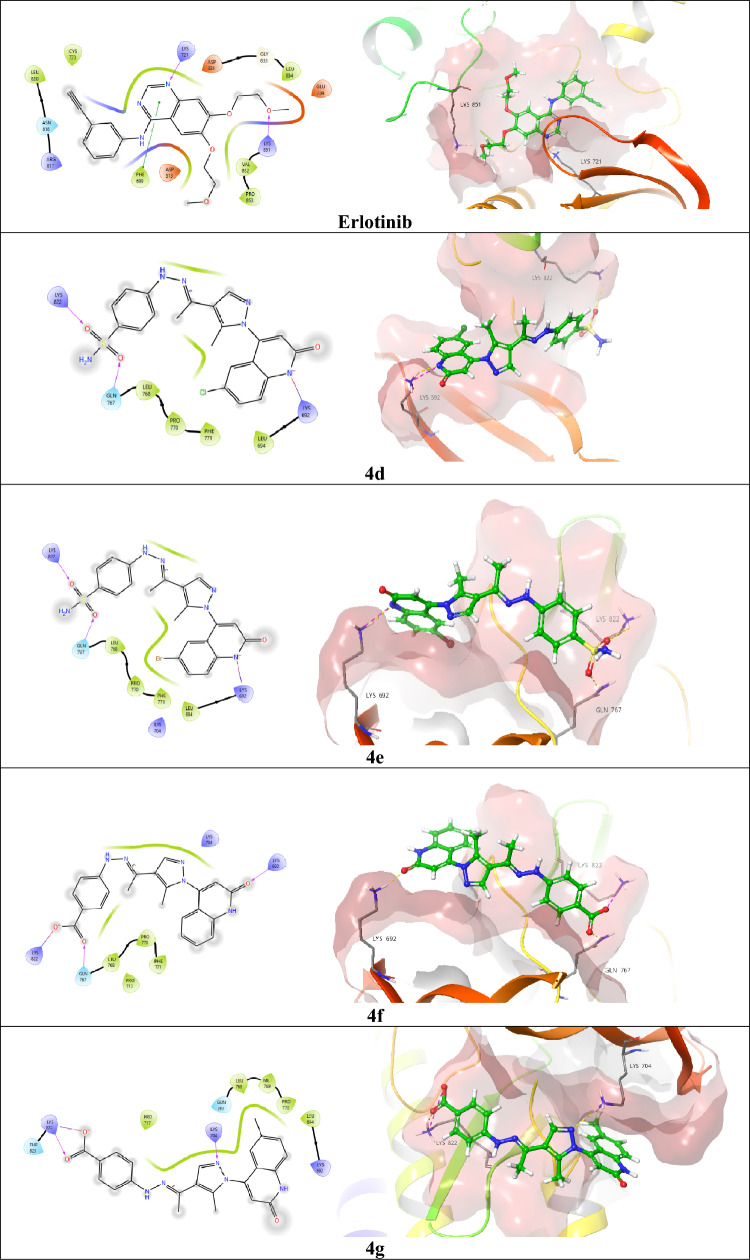 Figure 12
Figure 12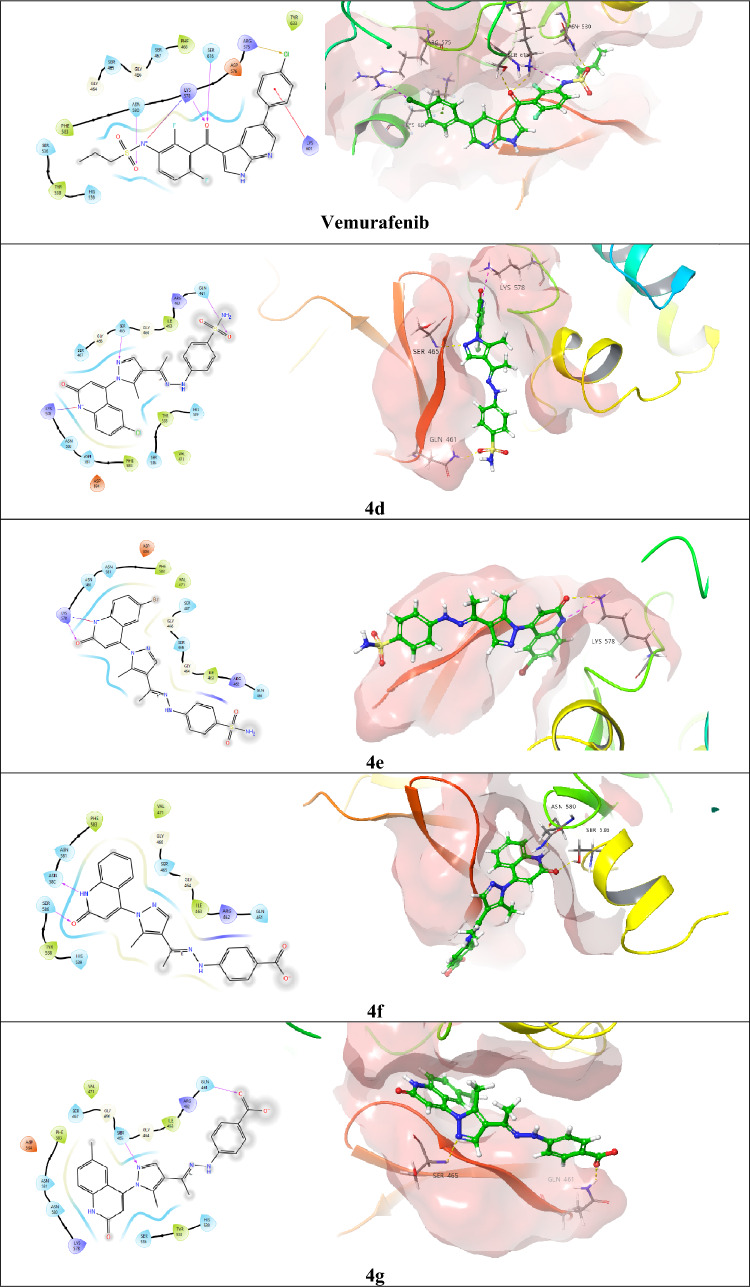 Figure 13
Figure 13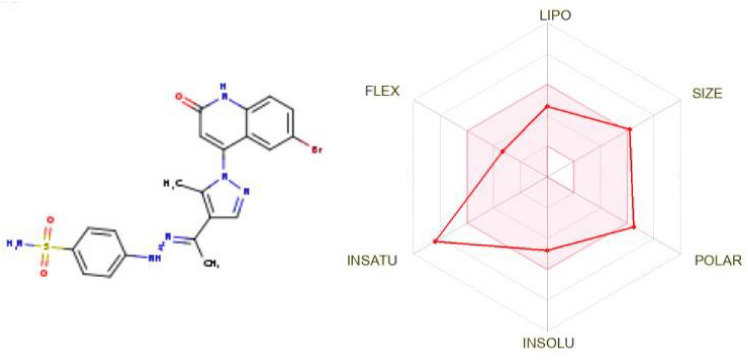 Figure 14
Figure 14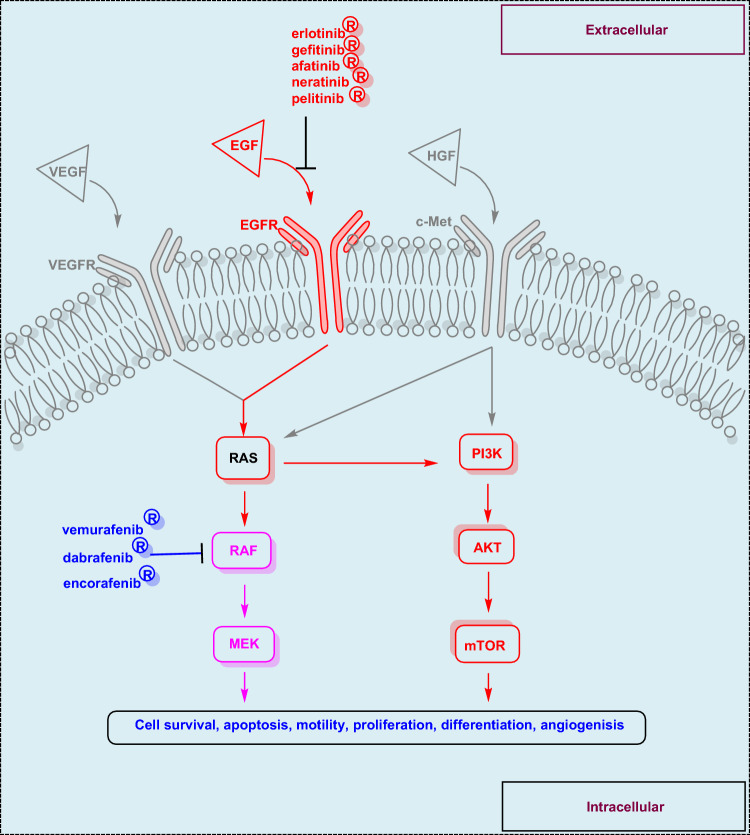 Figure 1
Figure 1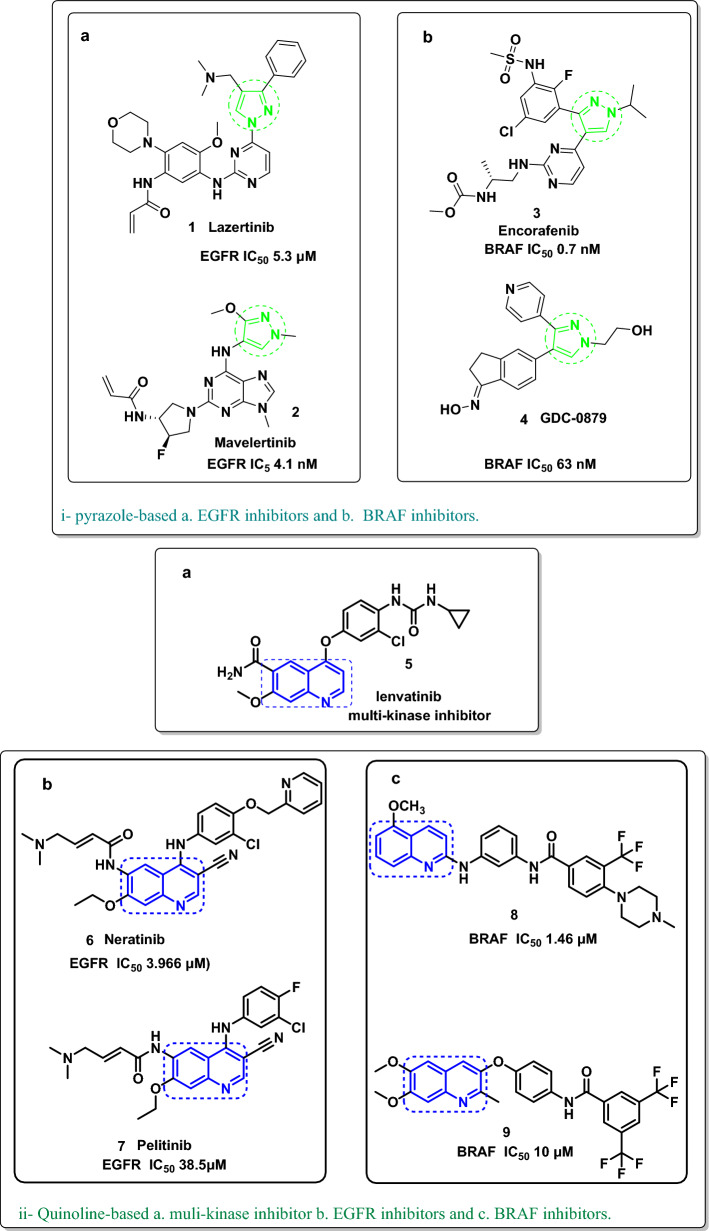 Figure 2
Figure 2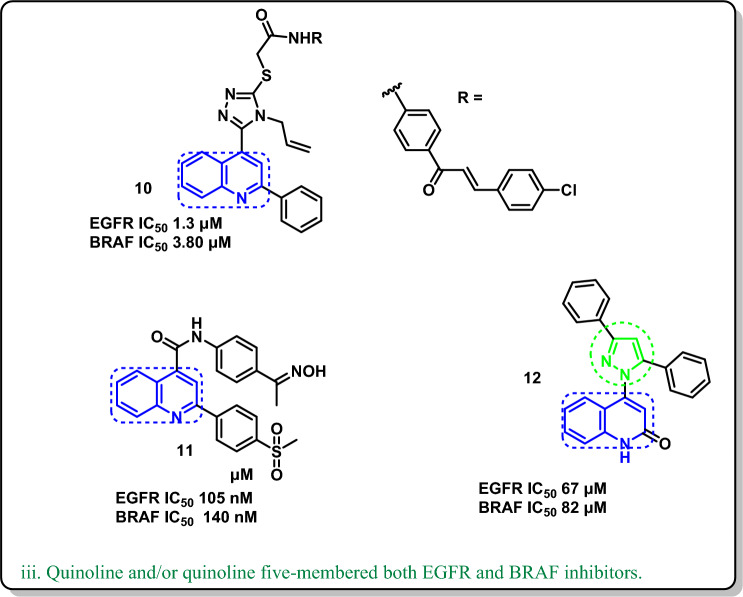 Figure 2
Figure 2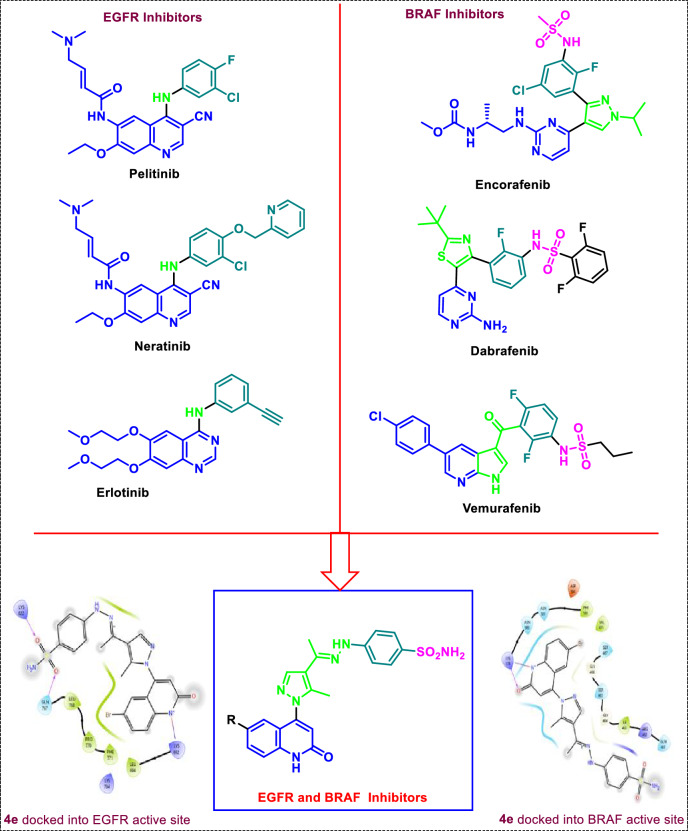 Figure 3
Figure 3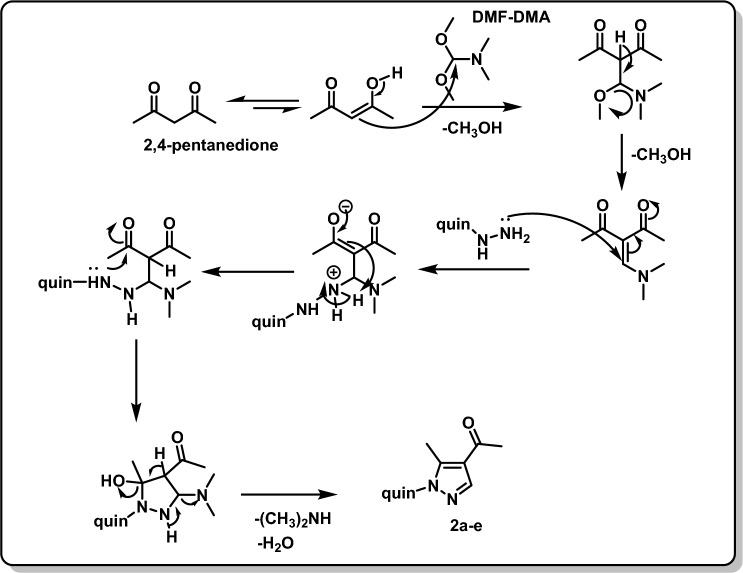 Figure 4
Figure 4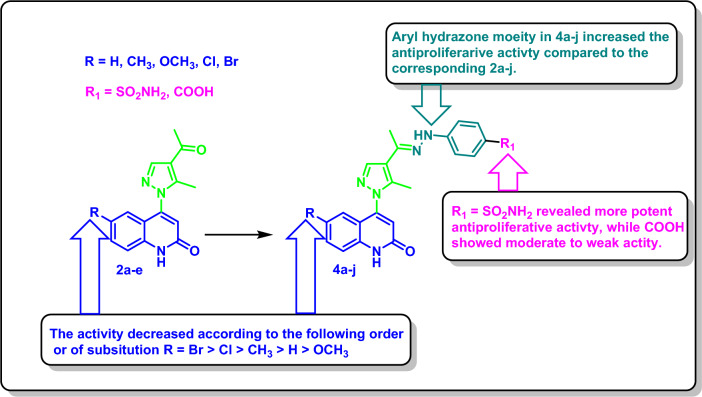 Figure 5
Figure 5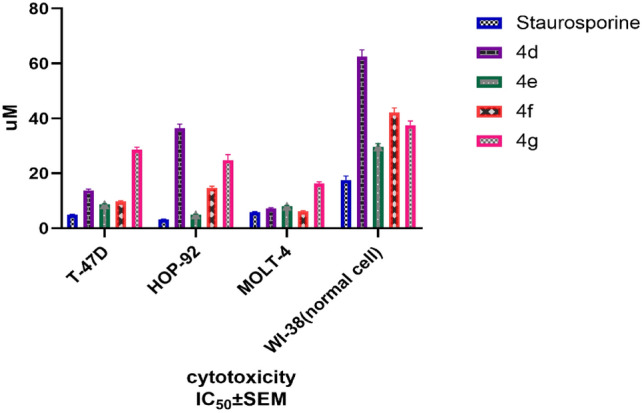 Figure 6
Figure 6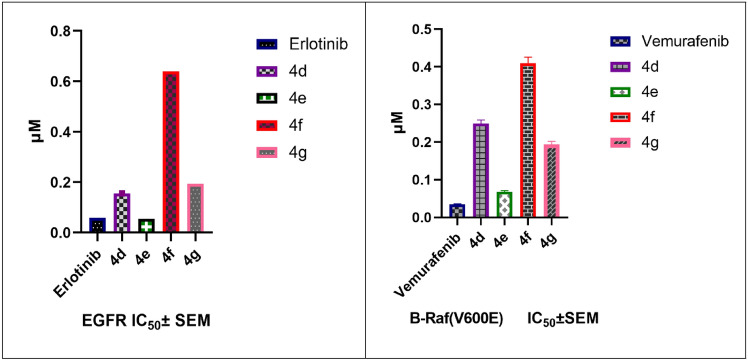 Figure 7
Figure 7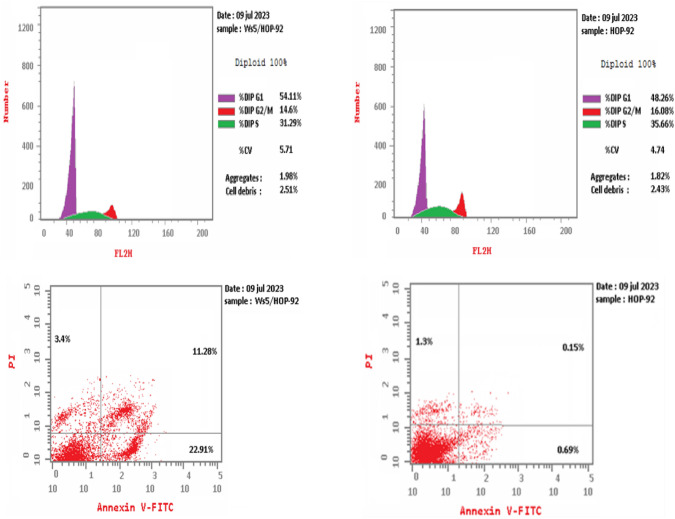 Figure 8
Figure 8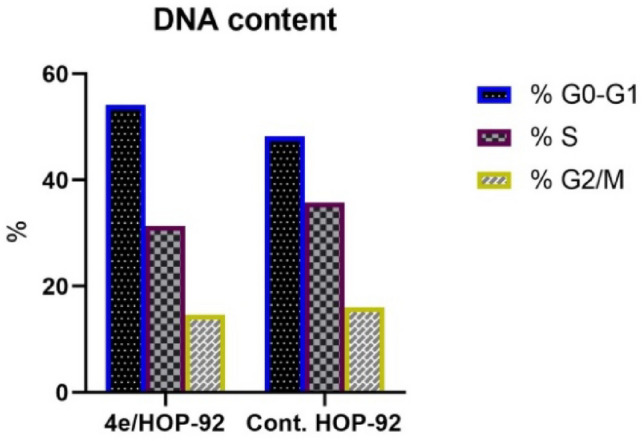 Figure 9
Figure 9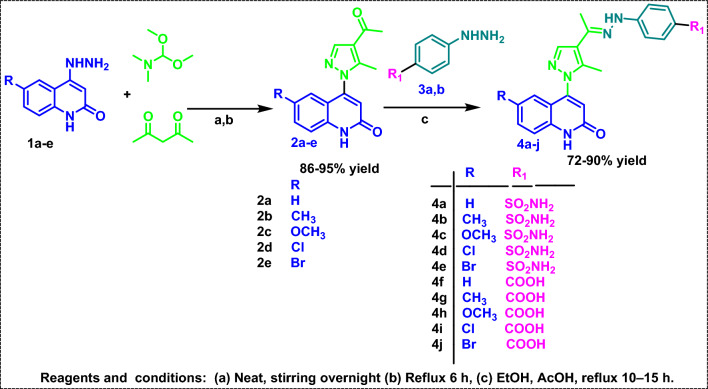 Figure 16
Figure 16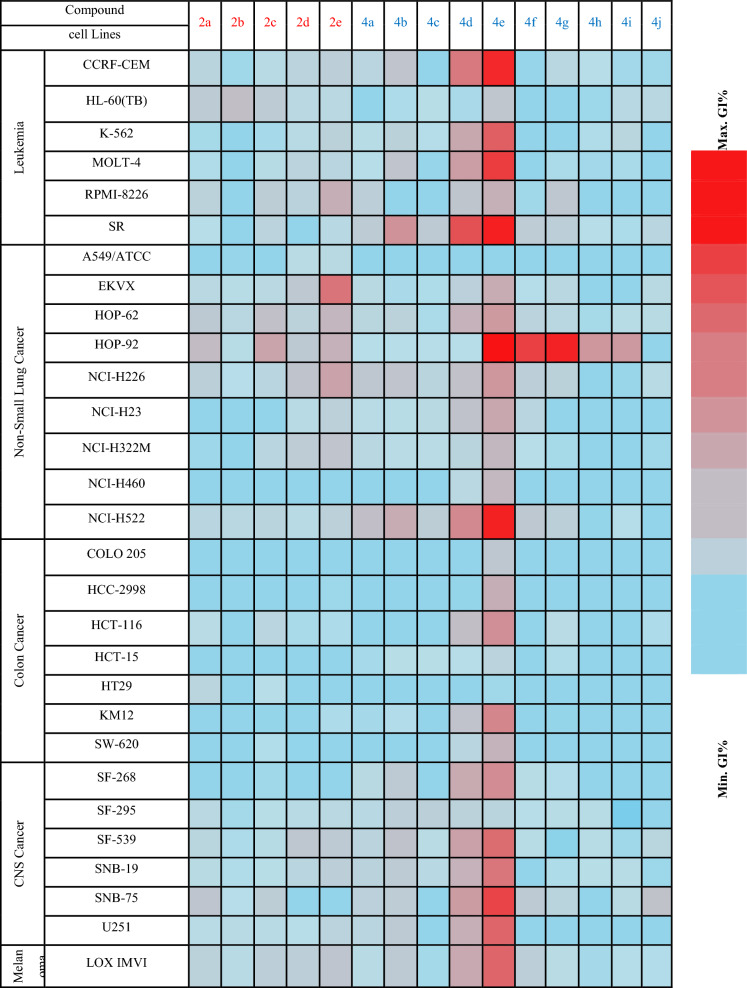 Figure 17
Figure 17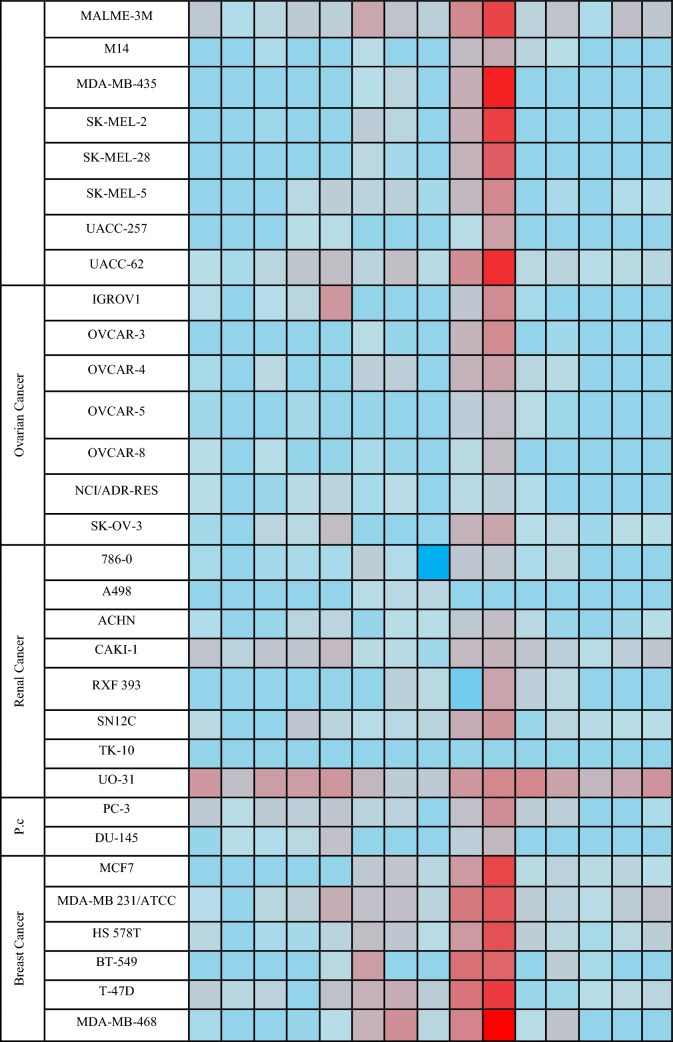 Figure 18
Figure 18- —Al-Azhar University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMelanoma and MAPK Pathways · Computational Drug Discovery Methods · Synthesis and biological activity
Introduction
Based on the revised projections from the International Agency for Research on Cancer (IARC), which breaks out worldwide cancer statistics by globe region for 2022, there were about 20 million new cases of cancer and 9.7 million cancer-related deaths [1]. Unfortunately, according to statistics, one in five men and women will have cancer at some point in their lives [2]. Biochemically, protein kinases are a family of enzymes that control a range of cellular biological processes, including apoptosis, proliferation, migration, metabolism, and finally normal cell growth and division [3]. Protein kinases regulate the previous biological process through interaction with its cell membrane-localized receptors, such as EGFR, VEGFR, c-Met, etc. [4]. Growth factors activate the EGFR, triggering a cascade of downstream waves within the cell that activate the RAS/RAF/MEK pathway. Consequently, the parallel pathways of PI3K/AKT/mTOR are stimulated by the activated intracellular RAS (Fig. 1) [5]. Overexpression, mutation, or disintegration of this tangled process results in uncontrolled cell division and proliferation [6]. Regarding cancer treatment strategy, blocking EGFR leads to RAS/RAF/MEK and PI3K/AKT/mTOR signaling cascade suppression [7]. In addition, BRAF^V600E^ is a significant intracellular protein that has recently been extensively studied as a distinct oncogenic element [8]. Unfavorable mutations in the BRAF^V600E^ gene accelerate tumor growth, leading to uncontrolled cell proliferation [7]. The known BRAF^V600E^ mutation was anticipated to serve as a resistance mechanism to much chemotherapy [9]. The development of resistance in cancer has also been linked to the feedback stimulation of EGFR signaling [10]. Hence, EGFR/BRAF (cetuximab/vemurafenib) was used to alleviate these issues according to studies on metastatic cases with BRAF^V600E^ mutations [11]. Therefore, the EGFR activation problem might be resolved by sequentially inhibiting the two kinases. One tactic for coincident blocking of more than one target is combined chemotherapy [12]. Unfortunately, dangerous drug interactions and toxicity can occur while taking several drugs concomitantly [13]. Therefore, combining two medications into a single molecule that hits several targets may help with these problems [14]. Based on the above-mentioned importance of EGFR and BRAF^V600E^ in controlling cell division and proliferation, dual targeting of EGFR and BRAF^V600E^ kinases revealed a successful strategy to control cancers [15]. The low efficacy, resistance, or toxicity related to many single-target or combination-based medicines were overcome by these dual mechanistic medications [16], which also had a more acceptable posology. Subsequently, investigating this dual mechanism could result in a treatment that works well and has fewer adverse effects.Fig. 1. Cell regulation due to EGFR, VEGFR, and c-Met downstream signals through RAS/RAF/MEK pathways
In this context, pyrazole derivatives (Lazertinib) 1 [17] (Mavelertinib) 2 [18] acted on EGFR as a single target (Fig. 2i-a); other pyrazole derivatives 3 (Encotafenib) [19] (GDC-9879) 4 [20] have the potential to inhibit BRAF^V600E^ (Fig. 2i-b). On the other side, quinoline in general is a wellknown as multi-kinase inhibitory scaffold such as in lenvatinib 5 which works by inhibiting several tyrosine kinase receptors, including VEGFR-1,2,3, FGFR-1,2,3,4, RET, and c-KIT [21]. Notably, the quinoline-bearing compounds (Neratinib) 6 [22] (Pelitinib) 7 [23] were published as EGFR inhibitors (Fig. 2ii-a). Moreover, the quinoline-derived 8 and 9 [24] revealed BRAF^V600E^ inhibitory activities (Fig. 2ii-b). In addition, the quinoline/triazole hybrid 10 [25] and the quinoline/amide-bridged 11 [26] showed promising dual inhibition of both the EGFR and BRAF^V600E^ oncogenic proteins. Further, as a possible twin inhibitory mechanism, the quinoline/pyrazole hybrid 12 [26] worked on both EGFR and BRAF^V600E^ targets (Fig. 2iii). Therefore, searching for a hybrid of quinoline and pyrazole may be the shortest way to obtain a dual-mechanism anticancer.Fig. 2. Quinoline/or quinoline five-membered ring EGFR/BRAF single and/or dual mechanism
Considering the Structure–activity relationship of the EGFR-TKIs [27] (Fig. 3); pelitinib, erlotinib [28], and neratinib could be summarized as follows: (a) the adenine pocket is occupied by a heteroaromatic scaffold; (b) a NH spacer that interacts with the amino acids inside the linker region; (c) a terminal hydrophobic moiety; and (d) a hydrophobic tail at the end that settles into the hydrophobic area. On the other hand, the requirements for BRAF^V600E^ inhibitors such as Encorafenib, Dabrafenib [29], and Vemurafenib [30] could be encapsulated in (a) a sulfonamide moiety or alkylated sulfonamide moiety which is essential for interacting with the important amino acid residue; (b) a phenyl moiety occupies the extended side of the pocket; (c) C=O, pyrazole, or imidazole linker; and (d) heteroaromatic system like azaindole or pyrimidine (bioisosteric quinoline as represented in our target compounds) which occupied the adenine pocket. Consequently, considering the essential requirements for EGFR and BRAF^V600E^ inhibitors and utilizing the bio versatility of both quinoline and pyrazole moieties, we designed a novel quinoline-pyrazole-based derivatives that have the main features and pharmacophores for both EGFR and BRAF^V600E^ inhibitors such as a quinoline heteroaromatic system, modified NH linker, or spacer with hydrazone attached to the pyrazole and phenyl sulfonamide moiety.Fig. 3. Rationale design for the final compounds 4a–j inspired from the reported EGFR and BRAF^V600E^-active-marketed anticancer drugs
Results and discussion
Chemistry
The intermediates 4-hydrazineylquinolin-2(1H)-one derivatives 1a–e were synthesized according to the reported method [31].
The final compounds have been synthesized using the procedures outlined in Scheme 1. An equimolar amount of 2,4-pentanedione and N,N-dimethylformamidedimethyl acetal (DMF-DMA) was stirred overnight. Then, 4-hydrazineylquinolin-2(1H)-ones 1a–e were added to form intermediates 2a–e according to the proposed Michael-addition mechanism (Fig. 4) [32]. The final compounds 4a–j were obtained by condensing acetyl pyrazolyl quinoline derivatives 2a–e with phenyl hydrazine derivatives 3a-b.Scheme 1. Synthetic approach to target compounds 4a–jFig. 4. The proposed Michael-addition mechanism for the synthesis of compounds 2a–e
Along with the expected compounds, confirmatory ^1^H NMR, ^13^C NMR, and HRMS analyses were confirmed. Compound 4e’s ^1^H NMR revealed a set of twelve protons between 9.61 and 6.78 δ ppm that were related to aromatic and sulfonamide protons, two methyl groups in the aliphatic region, and one proton at 12.29 δ ppm that corresponded to the NH amidic. In addition to the precise number of carbons, ^13^C NMR revealed two peaks in the aliphatic region that were ascribed to methyl carbons. Furthermore, compound 4e showed molecular ion peaks at m/z 515.0499, which corresponds to its molecular formula conforming its purity.
Biology
National Cancer Institute (NCI) screening
Ten final compounds 4a–j and five intermediates 2a–e have been selected by the National Cancer Institute (NCI) for anticancer screening over a panel of 60 cancer cells that cover a variety of histological tissues, including the central nervous system, leukemia, lung, colon, kidney, ovary, breast, and prostate, using the protocol established by the National Cancer Institute (NCI) [33]. In a primary screening, each compound was screened at a concentration of 10 µM. The results were displayed in the supplementary data and expressed as the mean GI% of the treated cells relative to the control cells [34]. The following heatmap (Table 1) highlighted that compounds 4d–g have promising activity on HOP-92 lung, MOLT-4 leukemia, and T-47D breast cell lines. Interestingly, compound 4e showed remarkable activity over the most NCI cancer cell lines.Table 1. Heatmap diagram of NCI-60 inhibition by compounds 2a–e and 4a–j at 10 µM*p.c. prostate cancer
Structure activity relationship (SAR)
The data on in vitro NCI screening findings (Table 1) show above points to the following main trend: (a) regarding to the electron-donating groups, quinolin-2-one’s activity was enhanced by halogen substitution on C-6. (b) The antiproliferative effect was more pronounced in the substituted phenyl hydrazone 4a–j than in the acetylated intermediates 2a–j. (c) Sulfonamide-substituted compounds 4a–e are more active compared to the carboxylated compounds 4f–j (Fig. 5).Fig. 5. Structure activity relationship (SAR) of 2a–e, and 4a–j
Antiproliferative assay
To a greater extent, the MTT assay [35] was employed to assess the antiproliferative effect of the previously screened promising hybrids 4d–g. The MTT assay results reassured the promising antiproliferative activity of compound 4e on the three human cancer cell lines: leukemia (MOLT-4), lung cancer (HOP-92), and breast cancer (T47D) cell lines with mean inhibitory concentration IC_50_ ± SEM 8.62 ± 0.34, 4.982 ± 0.2, and 8.023 ± 0.31 µM, respectively, in comparison to the Staurosporine reference [36] (a broad-spectrum protein kinase inhibitor), which has IC_50_ ± SEM µM 4.94 ± 0.19, 3.172 ± 0.19, and 5.856 ± 0.23, respectively, on the same cell lines. Moreover, compound 4e did not reveal significant cytotoxicity to the normal cell line WI-38, with IC_50_ ± SEM µM 29.62 ± 1.18 compared to the reference compound with IC_50_ ± SEM µM 17.54 ± 1.5 (Table 2, Fig. 6).Table 2. In vitro cytotoxicity of compounds 4d–g on MOLT-4, HOP-92, and T47D cancer cell lines and cytotoxicity on the normal WI-38 cell lineCompoundAntiproliferative activity IC_50_ ± SEM (µM)T-47DHOP-92MOLT-4W1-384d13.85 ± 0.4836.52 ± 1.467.24 ± 0.2962.46 ± 2.494e8.62 ± 0.344.982 ± 0.28.023 ± 0.3129.62 ± 1.184f28.56 ± 0.9924.93 ± 1.916.31 ± 0.6542.14 ± 1.684g9.71 ± 0.3414.72 ± 0.666.17 ± 0.2537.57 ± 1.5Staurosporine4.94 ± 0.193.172 ± 0.195.856 ± 0.2317.54 ± 1.5Fig. 6In vitro cytotoxicity of compounds 4d–g on MOLT-4, HOP-92, and T47D cancer cell lines and cytotoxicity on the normal WI-38 cell line
EGFR inhibitory activity
The inhibitory effect of the most potent antiproliferative derivatives 4d–g was examined against EGFR as a potential molecular target. The compounds’ IC_50_ values are shown in Table 3. The antiproliferative assay and this inhibitory assay produced identical results. As before, the most effective antiproliferative agent 4e (R = Br, R^1^ = SO_2_NH_2_) had an IC_50_ value of 0.055 ± 0.002 μM, which was much higher than the reference erlotinib IC_50_ of 0.06 ± 0.002 μM. According to these results, 4e may be a potential EGFR inhibitor with antiproliferative activity, which supports our previous theory on the mechanism of action of 4e (Table 3, Fig. 7). See Appendix B.Table 3. The IC_50_ values of compounds 4d–g against EGFR and BRAF^V600E^CompoundEGFR inhibitionBRAF^V600E^ inhibitionIC_50_ ± SEM (μM)IC_50_ ± SEM (μM)4d0.156 ± 0.0060.249 ± 0.014e0.055 ± 0.0020.068 ± 0.0034f0.64 ± 0.0230.410 ± 0.0164g0.194 ± 0.0070.194 ± 0.008Erlotinib0.06 ± 0.002NDVemurafenibND0.035 ± 0.001N.B: (ND) = Not DeterminedFig. 7The IC_50_ values of compounds 4d–g against EGFR and BRAF^V600E^
BRAFV600E inhibitory activity
Using Vemurafenib as the control medication, Compounds 4d–g were further assessed for their inhibitory activity against BRAF^V600E^. Compounds 4d–g showed moderate anti-BRAF^V600E^ activity (IC_50_ values ranging from 0.194 ± 0.008 to 0.410 ± 0.016 μM) compared to Vemurafenib (IC_50_ = 0.035 ± 0.001 μM). On the other hand, compound 4e, with an IC_50_ of 0.068 ± 0.003 μM, was the most potent inhibitor of BRAF^V600E^. These results imply that compound 4e may act with dual inhibition for EGFR and BRAF^V600E^ as an antiproliferative agent (Table 3, Fig. 7). See Appendix B.
Cell cycle analysis
Research has been done on how the most powerful antitumor compound, 4e, affects the HOP-92 cell cycle’s growth and apoptosis. HOP-92 cells were treated for 24 h at an IC_50_ value of 4e (4.982 μM). The results of the study (Table 4, Fig. 8) reveal that a higher apoptosis rate at the pre-G1 phase for compound 4e on HOP-92 with a percent of cell accumulation of 54.11% indicates that cell growth is arrested at the G1 phase. In S and G2/M phases, an unnoticeable change percent of cell accumulation was observed in HOP-92 treated with 4e (31.3%) and (14.6%), respectively (Table 4).Table 4. Cell cycle analysis of compound 4e in the HOP-92 cancer cell lineDNA contentCommentIC_50_ µM% G0–G1% S% G2/M4e/HOP-924.9854.1131.314.6Cell growth arrest at G1phaseCont. HOP-92–48.2635.716.08Fig. 8Cell cycle analysis and apoptosis detection of 4e in HOP-92 cancer cell line
Apoptosis assay
Using the Annexin V-FITC/PI test [37], the tendency of compound 4e to cause apoptosis and its potential association with cytotoxic action were evaluated. The cells were labeled with annexin V-FITC/PI and cultured for 24 h to detect any signs of apoptosis. The HOP-92 cell cycle analysis revealed that 4e treatment was followed by pre-G1 apoptotic signaling. According to research on early and late apoptosis and DNA content (Figs. 9, 10), 4e with 3.4% necrosis is probably going to result in a large amount of apoptosis (Table 5 and Fig. 10). Fig. 9DNA content of 4e in the HOP-92 cancer cell lineFig. 10Apoptosis detection of 4e in the HOP-92 cancer cell lineTable 5Apoptosis detection of 4e in the HOP-92 cancer cell lineConcApoptosisNecrosisIC_50_ μMTotalEarlyLate4e/HOP-924.98237.5922.9111.283.4Cont. HOP-92–2.140.690.151.3
Apoptotic indicators
In the depicted findings, compound 4e affected Bcl-2, caspase-3, and caspase-9, increasing the expression of caspase-3/9 and decreasing the expression of Bcl-2, confirming the apoptotic status [27]. Compound 4e was found to increase the levels of several mitochondrial apoptotic proteins, such as the main executioner protease (caspases-3/-9) and anti-apoptosis marker (Bcl-2). The results displayed in Fig. 11 showed that the levels of caspases-3 and -9 in HOP-92 cells treated with 4e were greater than those in the untreated control group by 4.716 and 3.082-fold, respectively (Table 6, Fig. 11). On the other hand, in comparison to the untreated reference, 4e administration of HOP-92 successfully decreased expression of the anti-apoptotic Bcl-2 protein to one third.Fig. 11. Effect of 4e on Bcl-2, Caspases-3, and Caspases-9 levels inside the HOP-92 cellsTable 6Effect of 4e on Bcl-2, Caspases-3, and Caspases-9 levels inside the HOP-92 cellsRT-PCR Fold ChangeFLDCodeIC_50_Casp-3Casp-9Bcl-24e/HOP-924.9824.7163.0820.299Staurosporine/HOP-923.1726.9065.5140.326Cont.HOP-92–111
In silico studies
Docking analyses of the most stable poses of quinolone-based hybrids 4d–g along with the selected reference ligands erlotinib and vemurafenib were accomplished against EGFR and BRAF^V600E^-active sites.
Molecular docking simulation of the quinolone-based hybrids 4d–g inside the EGFR-active site
Believing on, the EGFR kinase domain (EGFRK) adopts an NH2-terminal lobe (N-lobe) that is mainly composed of β-strands and one α-helix (αC), and larger COOH terminal lobe (C-lobe) that is typically α-helical. Moreover, researchers have discovered that a cleft, which divides the two lobes, combines ATP, ATP analogs, and ATP-competitive inhibitors [38, 39]. Also considering that, the ATP-binding pocket at the intermediate cleft is divided into three main binding regions: (1) The adenine region, or hinge region, where the adenine free amino group accommodates the H-bond donor effect with the Gln767 residue, and the adenine N1 position establishes H-bond acceptor effect with the neighboring Leu768 and Met769 residues, (2) The sugar region: where the ATP ribose is located and H-bonded by its hydroxyl groups with the Asp86 residue, and (3) the phosphate-binding region: where the triphosphate part of the ATP converges and is H-bonded to the crucial Lys33 and Asp145 amino acids [40, 41]. Consequently, all the reported EGFR inhibitors, such as 4-aminoquinazolines [42], pyrido[2,3-d]pyrimidines [43], and pyrrolo[2,3-b]pyridine [44, 45], have been counted as competitive inhibitors at the ATP-binding pocket, especially at the ATP-hinge region. Herein, we accomplished a molecular docking simulation of the reference drug erlotinib as a sort of 4-aminoquinazoline inhibitor [46] and the novel quinolone-based hybrids 4d–g to validate their relative affinity against the EGFR-active site. The reference erlotinib exerted a glide score of (S = − 2.485 kcal/mol) that could be attained through worthy H-bonding acceptor interactions of its ether oxygen with the Lys851 amino acid (1.83 A°) and the N1 position of the quinazoline ring with the Lys721 residue (2.31 A°). In addition to a precious pi-pi stacking with the Phe699 amino acid (4.35 A°). Fortunately, all the investigated quinolone-based hybrids 4d–g exhibited superior glide scoring than the reference drug erlotinib, whereas compound 4d (S = − 3.483 kcal/mol) utilizes its sulfonamide moiety to reveal two H-bond acceptor effects with Lys822 and the critical Gln767 amino acids, along with a prominent H-bond of the N1 position of the 2-quinolone ring with the key Lys692 (3.10 A°). Providently, comparable with the in vitro assay IC_50_ results, the sulfonamide-bearing compound 4e that accomplished the EGFR inhibitory effect of (0.055 μM) in Table 7 achieved glide scoring of (3.226 kcal/mol) through performing two H-bond acceptor interactions with the same residues Lys822 and Gln767 that interact with compound 4d. Nevertheless, it accommodates a valuable salt bridge with the crucial residue Lys692 (3.14 A°) (Table 7, Fig. 12). Also, the novel carboxy-bearing candidates 4f (S = − 4.407 kcal/mol) and 4g (S = − 3.806 kcal/mol) performed obvious salt bridges with the Lys822 residue due to their carboxylate oxygen, while compound 4f elucidated two H-bond acceptor interactions of the same bond length (1.82 A°) with the Lys692 and Gln767 residues due to its 2-quinolone carbonyl and carboxylate carbonyl, respectively.Table 7. Docking scores (kcal/mol) and the key involved residues in the interactions of the investigated compounds 4d, 4e, 4f, 4g, and the reference drug erlotinib, accompanied with their bond length values (A^○^) inside the EGFR-active site (PDB ID: 1M17)CompoundGlide score (kcal/ mol)H-bondH-bond length (A^○^)Salt bridgeSalt bridge length (A^○^)Pi-pi stackingPi-pi stacking length (A^○^)4d − 3.483Lys692 (1)3.10––––Gln767 (1)2.02Lys822 (1)2.474e − 3.226Gln767 (1)2.06Lys692 (1)3.14––Lys822 (1)2.574f − 4.407Lys692 (1)1.82Lys822 (1)4.26––Gln767 (1)1.824g − 3.806Lys704 (1)2.37Lys822 (1)4.62––Lys822 (1)1.66Erlotinib − 2.485Lys851 (1)1.83––Phe699 (1)4.35Lys721 (1)2.31Fig. 12Two-dimensional and three-dimensional (green ball and sticks) surface representation of the novel investigated quinolone-based hybrids 4d–g besides the reference compound erlotinib in the active site of EGFR
Meanwhile, compound 4g also clarified also two H-bond acceptor interactions with Lys704 (2.73 A°) and Lys822 (1.66 A°) due to the N2 of its pyrazole core and the carbonyl oxygen of its carboxylate, respectively (Table 7, Fig. 12). Wherefore, compound 4e-bearing pyrazole could be considered a promising EGFR inhibitor through ATP-competitive inhibition at the hinge region while it accommodates valuable contacts with Gln767 where the adenine part of ATP binds, and gaining a superior computational docking score and in vitro IC_50_ results over the reference erlotinib that requires further study.
Molecular docking simulation of the quinolone-based hybrids 4d–g inside the BRAFV600E-active site
As the BRAF^V600E^, within the mitogen-activated protein kinase (MAPK) signaling pathway, tends to be mutated in a significant number of cancers, particularly in about 50% of melanomas [47]. About 90% of BRAF^V600E^ homo- and heterodimer mutations correlated with cancer contain a distinct mutation of the Val600 residue to Glu600 (BRAF^V600E^), which prompts an active conformation without activation loop phosphorylation [48]. Therefore, approximately all patients promote drug resistance within about 6 months of treatment by BRAF^V600E^ inhibitors that predominantly arise through reactivation of the MAPK pathway that is called transactivation [49]. In the present work, we investigated vemurafenib reference drug [50] besides the quinolone-based hybrids 4d–g as chemically linked kinase inhibitors aiming to lock the BRAF^V600E^ dimers in an inactive conformation that cannot endure transactivation and, hence, confirm their potential against mutated instances (Table 8, Fig. 13). The reference compound vemurafenib elucidated a useful glide score of (− 3.266 kcal/mol), represented by four protruding H-bonding interactions with the Asn580, Arg575, Ser616, and Lys578 residues with bond lengths of 1.99, 2.33, 2.56, and 2.27 A°, respectively. In addition to a prominent salt bridge of sulfonamide nitrogen with the Lys578 amino acid and pi-cation interaction with the Lys601 besides the mutated Val600 residue. Fortunately, all the inspected quinolone-based hybrids 4d–g revealed better glide scoring than the reference vemurafenib. While compound 4d sulfonamide oxygen created an H-bond acceptor effect with the Gln461 residue (2.13 A°), along with another H-bond acceptor effect of the N_2_ of the pyrazole moiety with the Ser465 residue (2.43 A°), and a characteristic salt bridge of the 2-quinolone nitrogen with the crucial Lys578 residue to achieve a glide score of (− 4.045 kcal/mol). Moreover, the sulfonamide compound 4e that accomplished the most hopeful IC_50_ result among the investigated compounds (0.068 μM) displayed a glide score of (− 3.474 kcal/mol) through an obvious H-bond acceptor interaction of its 2-quinolone carbonyl oxygen with the key Lys578 and a prominent salt bridge of its 2-quinolone nitrogen with the same residue. Also, the carboxy compounds 4f and 4g presented superior glide scoring over the reference vemurafenib of (− 5.717 kcal/mol) and (− 4.815 kcal/mol), respectively. Whereas compound 4f demonstrated a useful H-bond donor effect of its 2-quinolone nitrogen with the Asn580 residue (2.21 A°), the 2-carbonyl oxygen protruded H-bond acceptor effect with the Ser536 amino acid (2.15 A°). In a diverse manner, compound 4g elucidated two H-bond acceptor effects due to its carboxylate carbonyl oxygen and N_2_ of the pyrazole core with Gln461 (1.95 A°) and Ser465 (2.53 A°), respectively. Lastly, the inspected quinolone-based hybrids 4d–g exposed superior binding affinity to the BRAF^V600E^-active site that exceeds the reference drug vemurafenib that may explain their capability to overcome the mutated consequences of the BRAF^V600E^ and treat the resistant malignant cases that require further investigation.Table 8. Docking scores (kcal/mol) and the key involved residues in the interactions of the investigated quinolone-based hybrids 4d–g and the reference drug vemurafenib, accompanied with their bond length values (A^○^) inside the BRAF^V600E^-active site (PDB ID: 5JRQ)CompoundGlide score (kcal/ mol)H-bondH-bond length (A^○^)Salt bridgeSalt bridge length (A^○^)Pi-cation interactionPi-cation bond length (A^○^)4d − 4.045Ser465 (1)2.43Lys578 (1)3.45––Gln461 (1)2.134e − 3.474Lys578 (1)2.66Lys578 (1)3.56––4f − 5.717Asn580 (1)2.21––––Ser536 (1)2.154g − 4.815Ser465 (1)2.53––––Gln461 (1)461Vemurafenib − 3.266Asn580 (1)1.99Lys578 (1)4.26Lys6016.51Arg575 (1)2.33Ser616 (1)2.56Lys578 (1)2.27Fig. 13Two-dimensional and three-dimensional (green ball and sticks) surface representation of the novel investigated quinolone-based hybrids 4d–g besides the reference compound vemurafenib in the active site of BRAF^V600E^
Pharmacokinetic profile (ADME) and drug-likeness
The online SwissADME tool [51] was used to calculate the expected physicochemical considerations. The obtained results provide a favorable impression of the unique synthesized candidates’ feasibility profiles. It seems that none of the produced compounds violate the Lipinski criteria. Pharmacokinetic analysis revealed that compounds 4d and 4e have limited gastrointestinal (GI) absorption, which is regarded as a crucial factor for oral delivery. Compounds 4f and 4g, on the other hand, had substantial GI absorption (Table 9, Fig. 14). The Synthetic accessibility (SA) value is crucial when examining medicinal chemistry; the values acquired increase in difficulty from 1 to 10, which they cannot surpass. Thankfully, it was discovered that every produced molecule had values between 3 and 4, with no compound approaching or surpassing 10. In summary, all the compounds have suitable physicochemical parameters to be drug candidates [52].Table 9. Predicted ADME parameters of the novel compounds 4d–4gComp.Physicochemical propertiesPharmacokineticsDrug likenessMedicinal chemistryHB A^a^HBD^b^TPSA^c^Log Po/w^d^LogS^e^GIALog Kp^f^LipinskiVio^g^SA4d63143.612.66−4.46Low−7.43Yes03.394e63143.612.74−4.73Low−7.65Yes13.414f53112.372.72−4.27High−6.77Yes03.234g53112.373.07−4.57High−6.60Yes03.36^a^: Number of hydrogen bond acceptors. ^b^: Number of hydrogen bond donors. ^c^: Topological polar surface area. ^d^: Partition coefficient (lipophilicity). ^e^: Water Solubility. ^f^:(skin permeation). ^g^: violationFig. 14Pharmacokinetic profile (ADME) and drug-likeness of compound 4e
Conclusion
A series of new compounds based on quinoline have been designed, synthesized, and tested for their ability to inhibit cell division. A panel of sixty cancer cell lines from the NCI was used to examine the newly produced compounds in vitro. Quinolone-based hybrids 4d–g showed encouraging antiproliferative activity in the MTT experiment when compared to Staurosporine, the positive control. Compound 4e was found to be the most effective; it may target EGFR and/or BRAF^V600E^ to function as an antiproliferative agent. Compound 4e displayed cell cycle arrest at the G0-1 phase and caused apoptosis against BRAF^V600E^ and EGFR, two potential dual targets for anticancer therapy.
Regarding the molecular docking studies, quinolone-based hybrids 4d–g clearly showed significant docking scores for EGFR and BRAF^V600E^ in relation to the ligands vemurafenib and erlotinib, respectively. Interestingly, compound 4e may prove to be a promising inhibitor for both the BRAF^V600E^ and EGFR targets following structural alteration.
Experimental
Chemistry
General details: See Appendix A
General synthetic procedure for 4-(4Aacetyl-5-methyl-1H-pyrazol-1-yl)-6-substituted quinolin-2(1H)-one derivatives. (2a–e)
A mixture of 2,4-Pentanedione (5.0 mmol) was mixed with N,N-dimethylformamide-dimethyl acetal (DMF-DMA) (6.0 mmol) and was stirred overnight at room temperature. Next, a 4-hydrazineylquinolin-2(1H)-one derivative (5 mmol) was added, and the reaction mixture was heated at 70–80 °C in ethanol for 6 h. The reaction mixture was subsequently allowed to cool and refrigerated for 24 h. Then the reaction mixture was filtrated and washed with ethanol.
4-(4-Acetyl-5-methyl-1H-pyrazol-1-yl)quinolin-2(1H)-one (2a)
Pale yellow powder, Yield: (93%); mp: 219–221 °C, ^1^H NMR (400 MHz, DMSO-d6) δ (ppm): 12.22 (1H s, 1H, NHCO), 8.38 (s, 1H), 7.60 (t, J = 7.7 Hz, 1H),7.44 (d, J = 8.2 Hz, 1H), 7.17 (t, J = 7.6 Hz, 1H), 6.96 (d, J = 7.5 Hz, 1H), 6.75 (s, 1H), 2.49 (s, 3H, –CH_3_), 2.43 (s, 3H, –CH_3_). ^13^C NMR (100 MHz, DMSO-d6) δ (ppm): 193.10, 161.29, 145.29, 144.03, 142.94, 139.33, 131.76, 123.99, 122.53, 120.62, 120.54, 116.36, 115.83, 28.85, 11.36. Anal. Calcd. For C_15_H_13_N_3_O_2_: C, 67.40; H, 4.90; N, 15.72. Found: C, 67.62; H, 5.06; N, 15.89.
4-(4-Acetyl-5-methyl-1H-pyrazol-1-yl)-6-methylquinolin-2(1H)-one (2b)
Yield: (88%); mp: 223–225 °C, ^1^H NMR (400 MHz, DMSO-d6) δ (ppm): 12.15 (s, 1H, NHCO), 8.38 (s, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 6.76 (s, 1H), 6.70 (s, 1H), 2.49 (s, 3H, –CH_3_), 2.42 (s, 3H, –CH_3_), 2.25 (s, 3H, –CH_3_ ^13^C NMR (100 MHz, DMSO-d6) δ (ppm): 193.36, 161.56, 145.51, 144.40, 143.42, 137.88, 133.50, 132.13, 123.65, 121.02, 120.94, 116.73, 116.26, 28.94, 20.88, 11.82. Anal. Calcd. For C_16_H_15_N_3_O_2_: C, 68.31; H, 5.37; N, 14.94. Found: C, 68.44; H, 5.49; N, 15.17.
4-(4-Acetyl-5-methyl-1H-pyrazol-1-yl)-6-methoxyquinolin-2(1H)-one (2c)
Yellow powder, Yield: (95%); mp: 211–213 °C, ^1^H NMR (400 MHz, DMSO-d6) δ (ppm): 12.13 (s, 1H, NHCO), 8.39 (s, 1H), 7.40 (d, J = 9.0 Hz, 1H), 7.30 (d, J = 8.9 Hz, 1H), 6.74 (s, 1H), 6.41 (s, 1H), 3.65 (s, 3H, OCH_3_), 2.49 (s, 3H, –CH_3_), 2.44 (s, 3H, –CH_3_). ^13^C NMR (100 MHz, DMSO-d6) δ (ppm): 193.74, 161.34, 155.01, 145.23, 144.53, 143.50, 134.31, 121.30, 121.11, 121.01, 117.82, 117.46, 106.21, 55.91, 29.28, 11.84. Anal. Calcd. For C_16_H_15_N_3_O_3_: C, 64.64; H, 5.09; N, 14.13. Found: C, 64.80; H, 5.21; N, 14.30.
4-(4Aacetyl-5-methyl-1H-pyrazol-1-yl)-6-chloroquinolin-2(1H)-one (2d)
Dark yellow powder, Yield: (91%); mp: 229–231 °C, ^1^H NMR (400 MHz, DMSO-d6) δ (ppm): 12.34 (s, 1H, NHCO), 8.41 (s, 1H), 7.66 (dd, J = 8.8, 2.4 Hz, 1H), 7.45 (d, J = 8.8 Hz, 1H), 7.03 (d, J = 2.3 Hz, 1H), 6.86 (s, 1H), 2.49 (s, 3H, –CH_3_), 2.47 (s, 3H, –CH_3_). ^13^C NMR (100 MHz, DMSO-d6) δ (ppm): 193.38, 161.17, 144.55, 144.15, 143.43, 138.30, 131.83, 126.62, 123.35, 121.64, 120.82, 118.04, 117.64, 28.89, 11.66. Anal. Calcd. For C_15_H_12_ClN_3_O_2_: C, 59.71; H, 4.01; Cl, 11.75; N, 13.93; Found: C, 59.89; H, 4.23; N, 14.15.
4-(4-Acetyl-5-methyl-1H-pyrazol-1-yl)-6-bromoquinolin-2(1H)-one (2e)
Brown powder, Yield: (86%); mp: 240–242 °C, ^1^H NMR (400 MHz, DMSO-d6) δ (ppm): 12.33 (s, 1H, NHCO), 8.41 (s, 1H), 7.76 (d, J = 8.8 Hz, 1H), 7.39 (d, J = 8.8 Hz, 1H), 7.16 (s, 1H), 6.84 (s, 1H), 2.49 (s, 3H), 2.47 (s, 3H). ^13^C NMR (100 MHz, DMSO-d6) δ (ppm): 193.53, 161.46, 144.85, 144.30, 143.74, 139.00, 134.74, 126.68, 121.94, 121.13, 118.52, 118.39, 114.24, 29.33, 12.11. Anal. Calcd. For C_15_H_12_BrN_3_O_2_: C, 52.04; H, 3.49. N, 12.14. Found: C, 52.16; H, 3.60; N, 12.37.
General synthetic procedure for (E)-4-(2-(1-(1-(6-substituted-2-oxo-1,2-dihydroquinolin-4-yl)-5-methyl-1H-pyrazol-4-yl) ethylidene)hydrazineyl)benzenesulfonamide derivatives. 4a–e and (E)-4-(2-(1-(1-(6-substituted-2-oxo-1,2-dihydroquinolin-4-yl)-5-methyl-1H-pyrazol-4-yl) ethylidene)hydrazineyl)benzoic acid derivatives. 4f–j
A sulfonamide phenylhydrazine derivative 3a or carboxylic phenylhydrazine derivative 3b (1.0 mmol) and 3 drops of glacial acetic acid were added to a suspension of compounds 2a–e (1.0 mmol) in ethanol (20 mL). The reaction mixture was refluxed for 10–12 h. The precipitate that formed after cooling was filtered out, washed with diethyl ether, and crystallized from 20 mL of ethanol.
(E)-4-(2-(1-(5-methyl-1-(2-oxo-1,2-dihydroquinolin-4-yl)-1H-pyrazol-4-yl)ethylidene) hydrazineyl)benzenesulfonamide (4a)
Pale yellow powder, yield: (76%); mp: 270–272 °C, ^1^H NMR (400 MHz, DMSO-d6) δ (ppm): 12.17 (s, 1H, NH), 9.59 (s, 1H), 8.09 (s, 1H), 7.68–7.53 (m, 3H), 7.44 (d, J = 8.2 Hz, 1H), 7.19 (m, 3H), 7.09–6.98 (m, 3H), 6.69 (s, 1H), 2.49 (s, 3H, –CH_3_), 2.32 (s, 3H, –CH_3_). ^13^C NMR (100 MHz, DMSO-d6) δ (ppm): 161.43, 148.78, 146.05, 140.24, 140.18, 139.32, 137.79, 133.04, 131.55, 127.25, 124.35, 122.37, 120.04, 116.74, 115.75, 111.57, 15.19, 12.59. HRMS (ESI, m/z) calcd for [C_21_H_21_N_6_O_3_S]^+^ (M + H)^+^ 437.1318, found. 437.1392.
(E)-4-(2-(1-(5-methyl-1-(6-methyl-2-oxo-1,2-dihydroquinolin-4-yl)-1H-pyrazol-4-yl) ethylidene)hydrazineyl)benzenesulfonamide (4b)
White powder, yield: (87%); mp: 289–291 °C, ^1^H NMR (400 MHz, DMSO-d6) δ (ppm): 12.11 (s, 1H, NHCO), 9.59 (s, 1H), 8.10 (s, 1H), 7.64 (d, J = 8.8 Hz, 2H), 7.40 (dd, J = 30.7, 8.5 Hz, 2H), 7.22 (d, J = 8.8 Hz, 2H), 7.03 (s, 2H), 6.86 (s, 1H), 6.65 (s, 1H), 2.50 (s, 3H), 2.33 (s, 3H), 2.27 (s, 3H). ^13^C NMR (100 MHz, DMSO-d6) δ (ppm): 161.06, 148.79, 145.82, 140.24, 137.71, 137.43, 133.03, 132.86, 131.46, 127.25, 123.58, 120.02, 119.95, 116.69, 115.74, 111.57, 20.41, 15.18, 12.61. HRMS (ESI, m/z) calcd for [C_22_H_23_N_6_O_3_S]^+^ (M + H)^+^ 451.1474, found. 451.1551.
(E)-4-(2-(1-(1-(6-methoxy-2-oxo-1,2-dihydroquinolin-4-yl)-5-methyl-1H-pyrazol-4-yl) ethylidene)hydrazineyl)benzenesulfonamide (4c)
White powder, yield: (77%); mp: 273–275 °C, ^1^H NMR (400 MHz, DMSO-d6) δ (ppm): 12.07 (s, 1H, NHCO), 9.58 (s, 1H), 8.10 (s, 1H), 7.63 (d, J = 8.1 Hz, 2H), 7.41–7.17 (m, 4H), 7.02 (s, 2H), 6.67 (s, 1H), 6.53 (s, 1H), 3.65 (s, 3H, –CH_3_), 2.49 (d, J = 5.0 Hz, 3H, –CH_3_), 2.31 (s, 3H –CH_3_). ^13^C NMR (100 MHz, DMSO-d6) δ (ppm): 161.42, 154.86, 149.25, 145.92, 140.76, 140.67, 138.25, 134.42, 133.51, 127.73, 120.84, 120.53, 117.83, 117.68, 112.05, 106.67, 55.89, 15.66, 13.13. HRMS (ESI, m/z) calcd for [C_22_H_231_N_6_O_4_S]^+^ (M + H)^+^ 467.1423, found. 467.1497.
(E)-4-(2-(1-(1-(6-chloro-2-oxo-1,2-dihydroquinolin-4-yl)-5-methyl-1H-pyrazol-4-yl) ethylidene)hydrazineyl)benzenesulfonamide(4d)
Pale yellow powder, yield: (74%); mp: 290–292 °C, ^1^H NMR (400 MHz, DMSO-d6) δ (ppm): 12.30 (s, 1H, NHCO), 9.60 (s, 1H), 8.13 (s, 1H), 7.65 (t, J = 7.9 Hz, 3H), 7.46 (d, J = 8.8 Hz, 1H), 7.22 (d, J = 8.8 Hz, 2H), 7.12 (s, 1H), 7.03 (s, 2H), 6.79 (s, 1H), 2.53 (s, 3H –CH_3_), 2.32 (s, 3H –CH_3_). ^13^C NMR (100 MHz, DMSO-d6) δ (ppm): 161.18, 148.75, 144.64, 140.60, 140.03, 138.22, 138.07, 133.08, 131.45, 127.25, 126.22, 123.57, 120.86, 120.29, 117.85, 117.80, 111.58, 15.20, 12.61. HRMS (ESI, m/z) calcd for [C_21_H_20_ClN_6_O_3_S]^+^ (M + H)^+^ 471.0928, found. 471.1002.
(E)-4-(2-(1-(1-(6-bromo-2-oxo-1,2-dihydroquinolin-4-yl)-5-methyl-1H-pyrazol-4-yl) ethylidene)hydrazineyl)benzenesulfonamide (4e)
Pale yellow powder, yield: (78%); mp: 293–295 °C, ^1^H NMR (400 MHz, DMSO-d6) δ (ppm): 12.29 (s, 1H, NHCO), 9.61 (s, 1H), 8.14 (s, 1H), 7.76 (dd, J = 8.8, 2.1 Hz, 1H), 7.64 (d, J = 8.8 Hz, 2H), 7.39 (d, J = 8.8 Hz, 1H), 7.24 (dd, J = 18.4, 5.5 Hz, 3H), 7.03 (s, 2H), 6.78 (s, 1H), 2.53 (s, 3H), 2.32 (s, 3H). ^13^C NMR (10 MHz, DMSO-d6) δ (ppm): 161.41, 148.91, 144.82, 140.78, 140.22, 138.63, 138.26, 134.30, 133.15, 127.42, 126.69, 120.88, 120.46, 118.46, 118.22, 114.21, 111.76, 15.32, 12.76. HRMS (ESI, m/z) calcd for [C_21_H_20_^79^BrN_6_O_3_S]^+^ (M + H)^+^ 515.0423, found. 515.0499.
(E)-4-(2-(1-(5-methyl-1-(2-oxo-1,2-dihydroquinolin-4-yl)-1H-pyrazol-4-yl)ethylidene) hydrazineyl)benzoic acid (4f)
Pale yellow powder, yield: (81%); mp: 268–270 °C, ^1^H NMR (400 MHz, DMSO-d6) δ (ppm): 12.16 (s, 2H, COOH, NHCO), 9.58 (s, 1H), 8.07 (s, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.57 (t, J = 7.4 Hz, 1H), 7.42 (d, J = 8.1 Hz, 1H), 7.15 (d, J = 8.3 Hz, 1H), 7.04 (d, J = 7.9 Hz, 1H), 6.68 (s, 1H), 2.47 (s, 1H), 2.30 (s, 1H). ^13^C NMR (100 MHz, DMSO-d6) δ (ppm): 167.39, 161.47, 149.81, 146.09, 140.27, 140.20, 139.32, 137.80, 131.56, 131.02, 124.36, 122.39, 120.08, 120.04, 119.90, 116.75, 115.77, 111.59, 15.26, 12.59. HRMS (ESI, m/z) calcd for [C_22_H_20_N_5_O_3_]^+^ (M + H)^+^ 402.1488, found. 402.1557.
(E)-4-(2-(1-(5-methyl-1-(6-methyl-2-oxo-1,2-dihydroquinolin-4-yl)-1H-pyrazol-4-yl) ethylidene)hydrazineyl)benzoic acid (4g)
Pale yellow powder, yield: (88%); mp: 255–257 °C, ^1^H NMR (400 MHz, DMSO-d6) δ (ppm): 12.20 (s, 1H, COOH), 12.10 (s, 1H, NHCO), 9.60 (s, 1H), 8.09 (s, 1H), 7.78 (d, J = 8.8 Hz, 2H), 7.42 (dd, J = 8.5, 1.7 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.17 (d, J = 8.8 Hz, 2H), 6.86 (s, 1H), 6.64 (s, 1H), 2.49 (s, 3H), 2.32 (s, 3H), 2.26 (s, 3H). ^13^C NMR (100 MHz, DMSO-d6) δ (ppm): 167.82, 161.74, 150.26, 146.29, 140.71, 140.69, 138.17, 137.89, 133.31, 131.92, 131.46, 124.05, 120.49, 120.43, 120.34, 117.15, 116.21, 112.04, 21.04, 15.72, 13.06. HRMS (ESI, m/z) calcd for [C_23_H_22_N_5_O_3_]^+^ (M + H)^+^ 416.1644, found. 416.1717.
(E)-4-(2-(1-(1-(6-methoxy-2-oxo-1,2-dihydroquinolin-4-yl)-5-methyl-1H-pyrazol-4-yl) ethylidene)hydrazineyl)benzoic acid (4h)
Pale yellow powder, yield: (90%); mp: 288–290 °C, ^1^H NMR (400 MHz, DMSO-d6) δ (ppm): 12.08 (s, 2H, COOH), 12.08 (s, 2H, NHCO), 9.59 (s, 1H), 8.10 (s, 1H), 7.78 (d, J = 8.8 Hz, 2H), 7.39 (d, J = 9.0 Hz, 1H), 7.28 (dd, J = 9.0, 2.7 Hz, 1H), 7.17 (d, J = 8.8 Hz, 2H), 6.67 (s, 1H), 6.53 (d, J = 2.7 Hz, 1H), 3.65 (s, 3H), 2.56–2.43 (m, 6H), 2.32 (s, 3H). ^13^C NMR (100 MHz, DMSO-d6) δ (ppm): 167.83, 161.43, 154.86, 150.26, 145.93, 140.76, 140.66, 138.25, 134.42, 131.47, 120.85, 120.81, 120.56, 120.35, 117.83, 117.68, 112.05, 106.67, 55.88, 40.60, 40.39, 40.18, 39.97, 39.76, 39.55, 39.34, 15.64, 13.12. HRMS (ESI, m/z) calcd for [C_23_H_22_N_5_O_4_]^+^ (M + H)^+^ 431.1594, found. 416.1666.
(E)-4-(2-(1-(1-(6-chloro-2-oxo-1,2-dihydroquinolin-4-yl)-5-methyl-1H-pyrazol-4-yl) ethylidene)hydrazineyl)benzoic acid (4i)
Pale yellow powder, yield: (78%); mp: 274–276 °C, ^1^H NMR (400 MHz, DMSO-d6) δ (ppm): 12.25 (s, 1H, COOH), 12.19 (s, 1H, s, 2H, COOH, NHCO), 9.58 (s, 1H), 8.09 (s, 1H), 7.75 (d, J = 7.8 Hz, 2H), 7.61 (d, J = 7.9 Hz, 1H), 7.42 (d, J = 8.5 Hz, 1H), 7.19–7.06 (m, 3H), 6.76 (s, 1H), 2.50 (s, 3H), 2.29 (s, 3H). ^13^C NMR (100 MHz, DMSO-d6) δ (ppm): 167.83, 161.66, 150.24, 145.12, 141.01, 140.50, 138.70, 138.53, 131.91, 131.47, 126.70, 124.06, 121.33, 120.78, 120.40, 118.32, 118.27, 112.06, 15.53, 13.09. HRMS (ESI, m/z) calcd for [C_22_H_19_^35^ClN_5_O_3_]^+^ (M + H)^+^ 436.1098, found. 416.1192.
(E)-4-(2-(1-(1-(6-bromo-2-oxo-1,2-dihydroquinolin-4-yl)-5-methyl-1H-pyrazol-4-yl) ethylidene)hydrazineyl)benzoic acid (4j)
Pale yellow powder, yield: (72%); mp: 281–283 °C, ^1^H NMR (400 MHz, DMSO-d6) δ (ppm): 12.29 (s, 2H, COOH), 12.29 (s, 2H, COOH, NHCO), 12.29 (s, 2H), 9.62 (s, 1H), 8.13 (s, 1H), 7.83–7.70 (m, 3H), 7.39 (d, J = 8.8 Hz, 1H), 7.27 (d, J = 2.1 Hz, 1H), 7.18 (d, J = 8.8 Hz, 2H), 6.78 (s, 1H), 2.54 (s, 3H), 2.33 (s, 3H). ^13^C NMR (100 MHz, DMSO-d6) δ (ppm): 167.83, 161.64, 150.23, 144.97, 141.09, 140.50, 139.01, 138.55, 134.56, 131.47, 127.04, 121.25, 120.78, 120.40, 118.77, 118.49, 114.42, 112.06, 15.65, 13.10. HRMS (ESI, m/z) calcd for [C_22_H_19_^79^BrN_3_O_3_]^+^ (M + H)^+^ 480.0593, found. 480.0669.
Biology
Antiproliferative assay
Using Staurosporine as a control, the MTT assay was utilized to assess the antiproliferative effect of hybrids 4d, 4e, 4f, and 4g versus three human cancer cell lines: the leukemia (MOLT-4) cell line, the lung cancer (HOP-92) cell line, and the breast cancer (T47D) cell line. The median inhibitory concentration (IC_50_) and GI_50_ (average IC_50_) against the three cancer cell lines were calculated. See Appendix B.
EGFR inhibitory activity
The most potent Antiproliferative derivatives, 4d–g, were tested for inhibitory effect against EGFR as a possible molecular target. Assessed using a 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazol (MTT) test. For more details, see Appendix B.
BRAFV600E inhibitory assay
Using Vemurafenib as the control medication, Compounds 4d, 4e, 4f, and 4g were further assessed for their inhibitory activity against BRAF^V600E^. Refer to Appendix B for further information.
Docking study
Docking analyses of the most stable poses of 2-quinolone candidates incorporating pyrazole core 4d, 4e, 4f, and 4g along with the selected reference ligands: erlotinib and vemurafenib were accomplished against EGFR- and BRAF-active sites using Maestro software of Schrodinger 2022.4. The nominated biotargets are (1) EGFR (PDB ID: 1M17) [53] and (2) BRAF (PDB ID:5JRQ) [54] that were regained from RCSB protein data bank (PDB). The most stable conformers of the appointed 2-quinolones compounds and the biotargets reference ligands were built using ChemBiodraw 18.0 PerkinElmer software and cleaned up for bond alignment then imported with Schrodinger software in a 3D demonstration mode. The energy minimization has occurred using OPLS3e (Optimized Potentials for Liquid Simulations) [55] force field in Ligprep (Version 2022-4, Schrodinger) [56]. This minimization aids to assign bond orders, the hydrogens addition to the selected ligands, then the generated output files that denote the best poses of the ligands, were further utilized for docking analyses. Furthermore, the indicated targets were displayed for preparation using Protein preparation wizard (Version 2022-4, Schrodinger) [56] as a main tool for protein preparation. The hydrogens have been added to the proteins, the charges, and the produced Het states using Epik at pH 7.0 ± 2.0 were allocated. The targets have been exposed to Pre-process, protein modification, and refinement by the applicable chain selection and the water molecules have been removed. Lastly, the targets have been minimized using OPLS3 force field. Besides, the receptor grids were constructed by considering the co-crystal ligands (X-ray pose of the ligand in the protein). The centroid of the ligands has been selected to establish a grid box around it and VanderWaal radius of the target atoms was scaled to 1.00 Å with a partial atomic charge of 0.25. The molecular docking has been run using performed glide rigid docking protocol [57] by means of Maestro software of Schrodinger. All the docking estimates have been done using Extra Precision (XP) mode, where a scaling factor of 0.8 and a partial atomic charge < 0.15 were employed to the atoms of proteins. Glide docking scoring has been devoted for verifying the best-docked poses from the output, and the interactions of these docked poses have been further investigated using XP visualizer. After the last step of docking with the co-crystal ligand in XP mode, root-mean-square deviation (RMSD) was ensured to authorize the target, and the RMSD values lie within the range of 0.46 Å.
Supplementary Information
Below is the link to the electronic supplementary material.Supplementary file1 (DOCX 28806 KB)Supplementary file2 (DOCX 32920 KB)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Shiri P, Ramezanpour S, Amani AM, Dehaen W (2022) A patent review on efficient strategies for the total synthesis of pazopanib, regorafenib and lenvatinib as novel anti-angiogenesis receptor tyrosine kinase inhibitors for cancer therapy. Mol Divers 26(5):2981–3002. 10.1007/s 11030-022-10406-810.1007/s 11030-022-10406-835235141 · doi ↗ · pubmed ↗
- 2Ghasemi M, Liang S, Luu QM, Kempson I (2023) The MTT assay: a method for error minimization and interpretation in measuring cytotoxicity and estimating cell viability. In: Cell viability assays: methods and protocols. Springer, New York, pp 15–33. 10.1007/978-1-0716-3052-5_210.1007/978-1-0716-3052-5_237142913 · doi ↗ · pubmed ↗
- 3Heinrich T, Seenisamy J, Emmanuvel L, Kulkarni SS, Bomke J, Rohdich F, Greiner H, Esdar C, Krier M, Grädler U (2013) Fragment-based discovery of new highly substituted 1 H-pyrrolo [2,3-b]-and 3 H-imidazolo [4,5-b]-pyridines as focal adhesion kinase inhibitors. J Med Chem 56(3):1160–1170. 10.1021/jm 301601410.1021/jm 301601423294348 · doi ↗ · pubmed ↗