Hepatitis C Virus NS5A Inhibits YAP1-HSC70 Interaction, Thereby Preventing YAP1 Degradation Via Chaperone-Mediated Autophagy
MARIA ALETHEA SEPTIANASTITI, CHIEKO MATSUI, ZIHAN XU, FRANSISCA PUSPITASARI, LIN DENG, TAKAYUKI ABE, IKUO SHOJI

TL;DR
This study shows how the Hepatitis C virus prevents the degradation of a protein called YAP1, which may promote cell survival and disease progression.
Contribution
The study identifies a KFERQ motif in YAP1 and shows that HCV NS5A inhibits its degradation via chaperone-mediated autophagy.
Findings
YAP1 contains a KFERQ motif (324ELLRQ328) that binds to HSC70, a key component of chaperone-mediated autophagy.
HCV NS5A disrupts the interaction between YAP1 and HSC70, preventing YAP1 degradation and promoting its nuclear translocation.
Knockdown of LAMP2A, a CMA receptor, increases YAP1 levels, confirming YAP1 as a CMA substrate.
Abstract
Hepatitis C virus (HCV) infection induces chaperone-mediated autophagy (CMA), where the HCV NS5A protein promotes the binding of the molecular chaperone HSC70 to substrate proteins containing a KFERQ motif. HSC70 recognizes this pentapeptide motif and transports substrates to lysosomes for degradation. In this study, we identified a KFERQ motif (324ELLRQ328) in the YAP1 protein, a key regulator of the Hippo pathway, and examined its interaction with HSC70 during HCV infection. To determine whether HSC70 directly binds to this motif, we generated four YAP1 mutants with specific alterations in the KFERQ motif and assessed their binding to HSC70. Among these, the R327A/Q328A mutant showed the greatest reduction in HSC70 binding, indicating that the sequence 324ELLRQ328 functions as the KFERQ motif in YAP1. Further analysis revealed that YAP1 binds to the substrate-binding domain of HSC70.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
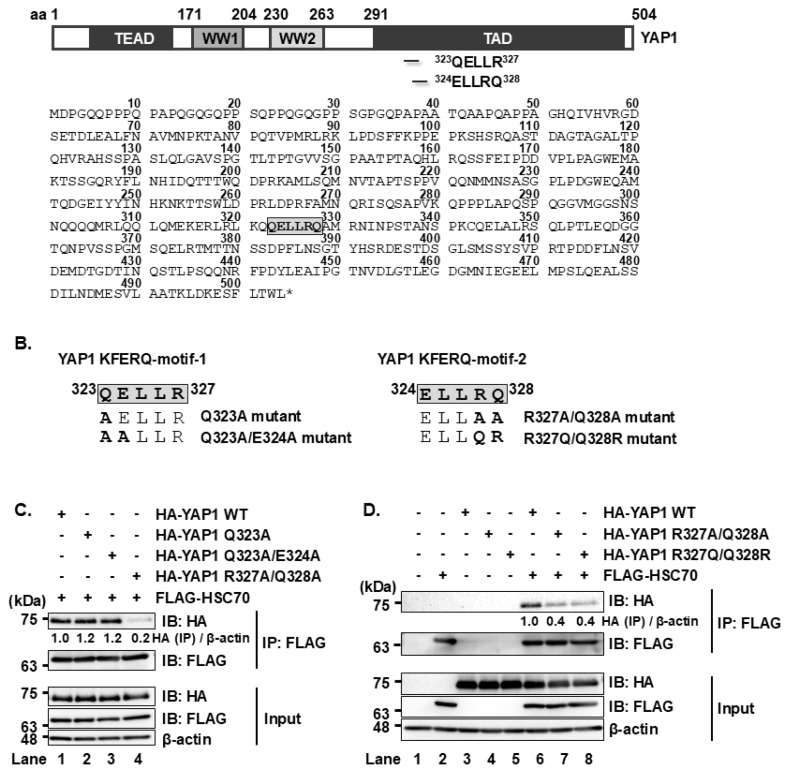 Figure 1
Figure 1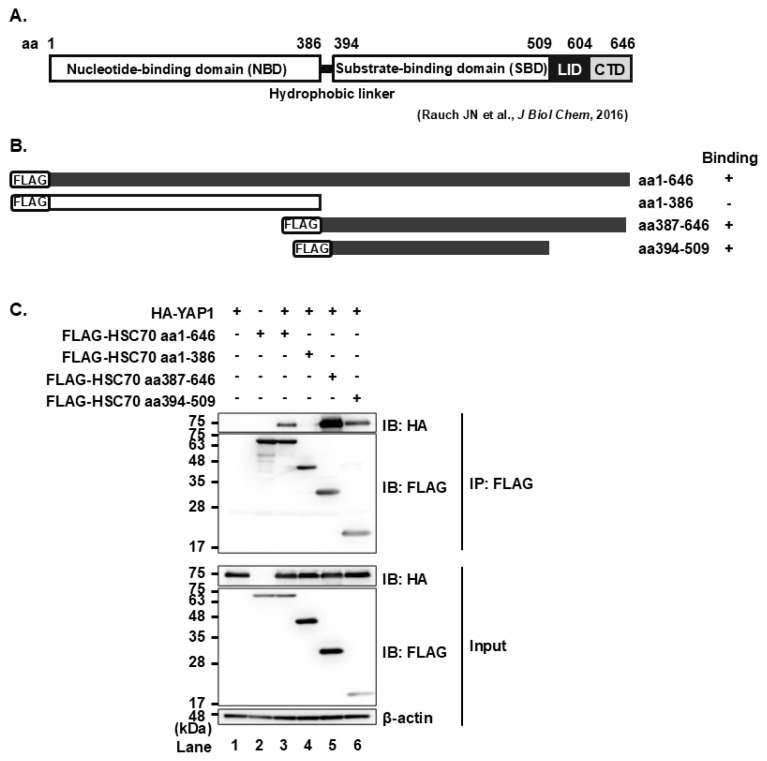 Figure 2
Figure 2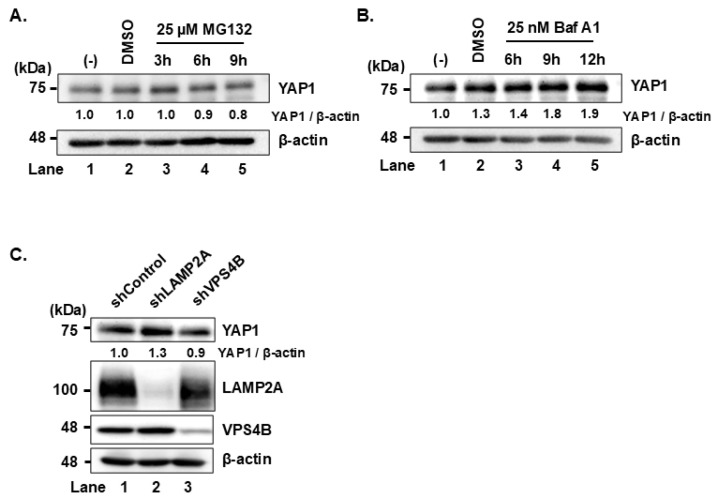 Figure 3
Figure 3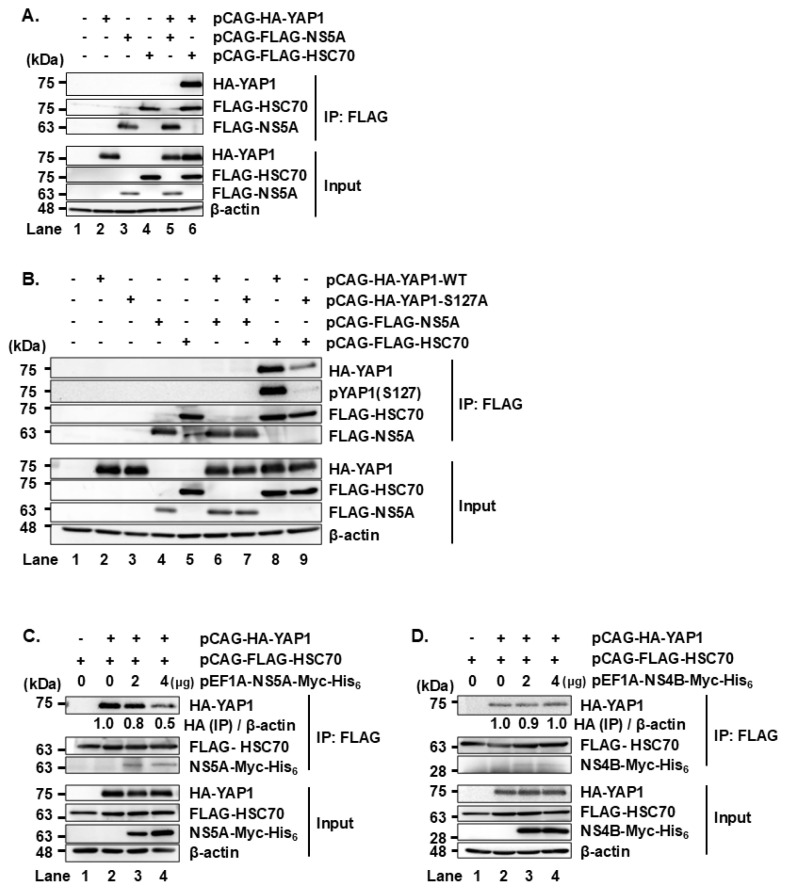 Figure 4
Figure 4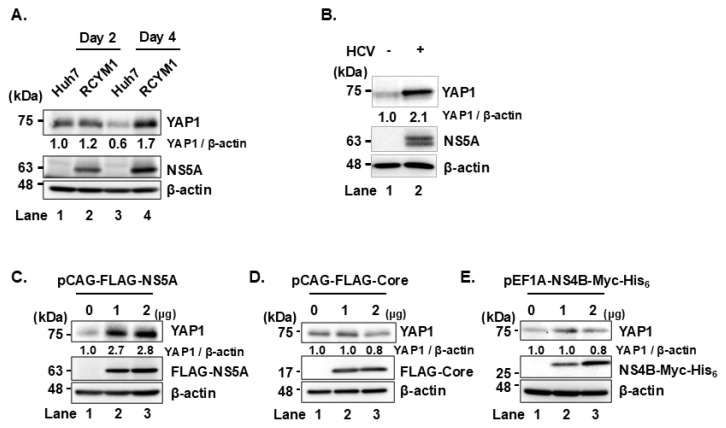 Figure 5
Figure 5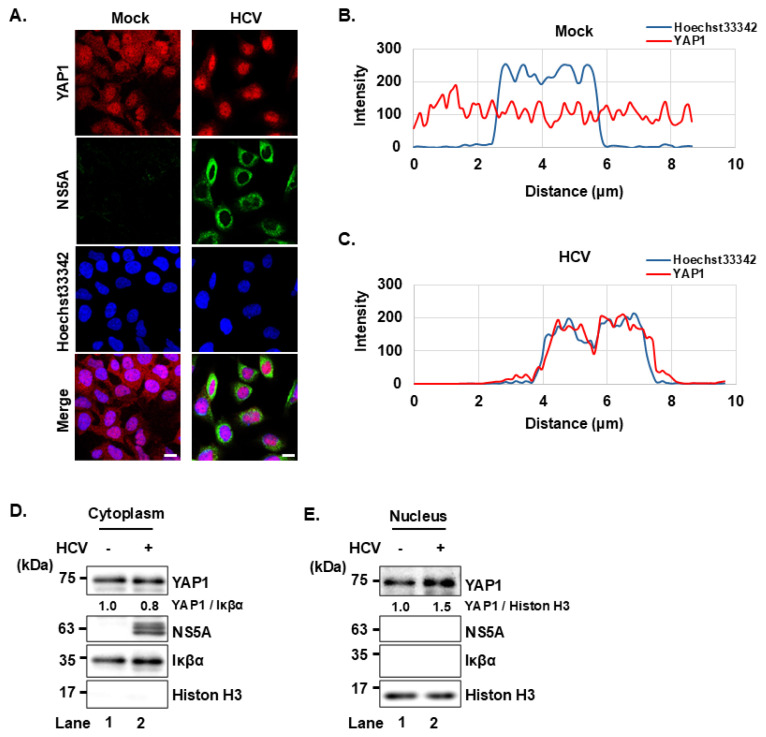 Figure 6
Figure 6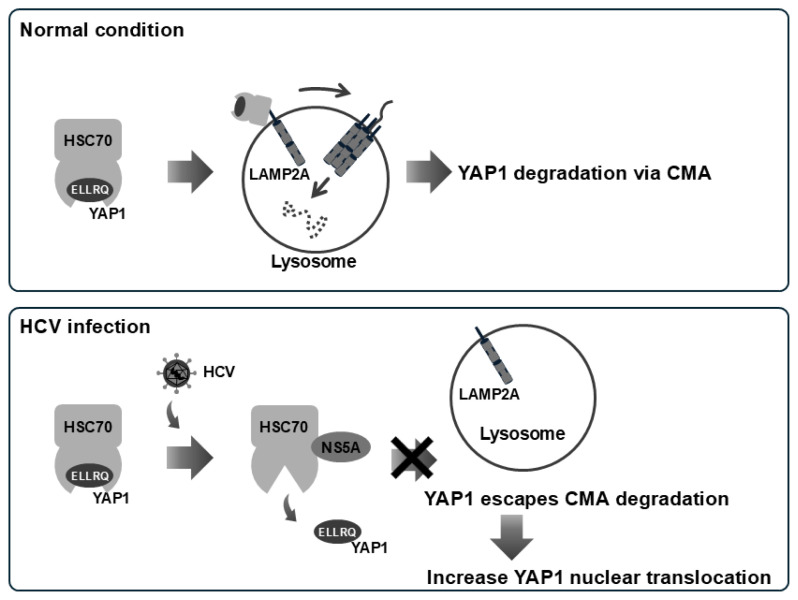 Figure 7
Figure 7- —Basic and Clinical Research on Hepatitis from the Japan Agency for Medical Research and Development (AMED)
- —KAKENHI
- —Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHippo pathway signaling and YAP/TAZ · Autophagy in Disease and Therapy · Cell death mechanisms and regulation
INTRODUCTION
Many viruses, including hepatitis C virus (HCV), exploit cellular protein degradation systems to promote viral propagation and pathogenesis. Autophagy is a key cellular process that delivers cytoplasmic components to lysosomes for degradation. While chaperone-mediated autophagy (CMA) was once considered to be the only selective lysosomal autophagy pathway, recent studies have identified endosomal microautophagy (eMI) as another pathway capable of selective substrate degradation [1].
We previously demonstrated that HCV enhances CMA by promoting the interaction between the cellular chaperone HSC70 and the transcription factor HNF-1α via the HCV NS5A protein, leading to lysosomal degradation of HNF-1α and subsequent downregulation of glucose transporter 2 expression [2–5]. Additionally, we found that HCV infection augments eMI by facilitating the interaction between HSC70 and diacylglycerol O-acyltransferase 1 (DGAT1) through NS5A, resulting in the selective degradation of DGAT1 [6]. Despite these findings, the broader mechanisms by which HCV exploit selective lysosomal degradation pathways, such as CMA and eMI, and their implications in viral life cycle and disease progression remain largely unclear.
The substrate selectivity in these pathways governed by the presence of a specific pentapeptide sequence, known as the KFERQ motif, in the target protein [7, 8]. The KFERQ motif typically includes one or two positively charged residues (lysine [K] and arginine [R]); one or two hydrophobic residues (phenylalanine [F], isoleucine [I], leucine [L], and valine [V]); one negatively charged residues (aspartic acid [D] or glutamic acid [E]); and one glutamine (Q) located on either side of the pentapeptide [9–11]. Cuervo and colleagues have developed a freely accessible web tool, KFERQ finder V0.8 (https://rshine.einsteinmed.edu/), to identify such motifs in protein sequences [12].
The interaction between HSC70 and the KFERQ-motif containing proteins is crucial for both CMA and eMI. In CMA, HSC70 recognizes the KFERQ motif and delivers the substrate to LAMP2A on the lysosomal membrane, where the substrate is unfolded and translocated for degradation. On the other hand, eMI involves the sequestration of substrate proteins within the late endosomal membrane. Following substrate binding, HSC70 interacts with phosphatidylserine (PS) on the endosomal membrane, triggering membrane invagination. The process is mediated by tumor susceptibility gene (TSG) 101, a component of endosomal sorting complex required for transport (ESCRT I), along with vacuolar protein sorting-associated protein (VPS) 4A, VPS4B, and Alix [13–15].
HSC70 is a 70-kDa ATP-dependent molecular chaperone that plays a central role in various cellular processes, including the prevention of protein misfolding and aggregation under both normal and stressed conditions [16]. Structurally, HSC70 consists of a nucleotide-binding domain (NBD) and a substrate-binding domain (SBD) [17]. The SBD is further subdivided into a β-sandwich subdomain containing a hydrophobic cleft for substrate binding [18], a helical LID subdomain that regulates access to the cleft and a disordered C-terminal domain (CTD), which serves as a site of post-translational modification and protein-protein interaction [19, 20].
The transcription factor Yes-associated protein 1 (YAP1) is a key effector of Hippo pathway and plays a critical role in liver pathogenesis by promoting cell proliferation, cell survival, migration, and metastasis [21–23]. When the Hippo pathway is active, YAP1 is phosphorylated at serine 127, enabling its interaction with 14-3-3 proteins and resulting in cytoplasmic retention. In contrast, when the Hippo pathway is inactivated, unphosphorylated YAP1 translocates to the nucleus, where it activates transcription of genes involved in cell proliferation. Recent studies have shown that YAP1 is a CMA substrate in HuS, Hep3B, and HepG2 cells. Impaired degradation of YAP1 via CMA has been linked to increased proliferation and migration in both normal and hepatocellular carcinoma cells [24].
We previously demonstrated that HCV NS5A facilitates CMA and eMI by enhancing HSC70-substrate interactions. In this study, we identified a KFERQ motif within the YAP1 protein sequence, suggesting that YAP1 is a potential substrate for CMA-mediated degradation. Thus, we investigated whether YAP1 is targeted for degradation via HCV-induced CMA or eMI. Our results showed that NS5A did not directly bind YAP1. Instead, NS5A disrupts the interaction between HSC70 and YAP1, thereby preventing YAP1 degradation via CMA. Since increased YAP1 activity is linked to increased cell proliferation and migration, the ability of HCV to protect YAP1 from CMA-mediated degradation may represent a novel mechanism underlying HCV-induced pathogenesis.
MATERIALS AND METHODS
Cell culture and viruses
The human hepatoma cell line, Huh-7.5 cells, was provided by Dr. Charles M. Rice (The Rockefeller University, New York, NY) [25]. Cells were maintained in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with L-glutamine and phenol red (044-29765; Fuji Film Wako Pure Chemical Industries, Osaka, Japan), 50 IU/ml penicillin, 50 μg/ml streptomycin (15-140-122; Gibco, Grand Island, NY), 10% heat-inactivated fetal bovine serum (S1760-500; Biowest, Nuaillé, France), and 0.1 mM non-essential amino acids (Invitrogen, Carlsbad, CA). Cells were incubated at 37°C in a 5% CO_2_ incubator. Phosphate-buffered saline (PBS) without calcium and magnesium (PBS [–]; 05913; Nissui Pharmaceutical Co., Ltd., Tokyo, Japan) was used for washing. Transfections were carried out using FuGENE 6 transfection reagents (E269A; Promega, Madison, WI). The pFL-J6/JFH1 plasmid encoding the entire viral genome of a chimeric strain of HCV-2a, J6/JFH1 was kindly provided by Dr. C.M. Rice [26]. The HCV genome RNA was synthesized in vitro using pFL-J6/JFH1 as a template and was transfected into Huh-7.5 cells by electroporation [27–29]. The virus produced in the culture supernatant was used for infection experiments [28]. Huh-7 cells stably harboring the HCV-1b full-genome replicon (FGR) derived from Con1 (RCYM1) were also utilized [30].
Expression plasmids
Expression plasmids encoding FLAG-tagged or Myc-His_6_-tagged NS5A and its deletion mutants have been previously described [2–4, 6, 31, 32]. The plasmid expressing HSC70 was also described previously [2, 6]. To clone YAP1, total RNA was extracted from Huh-7.5 cells and reverse-transcribed, followed by PCR amplification using the following primers:
Forward: 5′-TCGAGCTCAGCGGCCGCCATGGATCCCGGGCAGCAG-3′Reverse: 5′-AGTGAATTCGCGGCCGCTTATAACCATGTAAGAAA-3′
The amplified PCR product was purified and inserted into NotI site of pCAG-HA vector using the In-Fusion HD cloning kit (Takara Bio USA, Inc., 639649). Point mutations in YAP1 were introduced by overlap extension PCR using pCAG-HA-YAP1 as a template. Primer sequences used to generate the mutants were as follows:
Q323A:
Forward: 5′-CGGCTGAAACAGGCAGAACTGCTTCGGCAG-3′Reverse: 5′-CTGCCGAAGCAGTTCTGCCTGTTTCAGCCG-3′
Q323A/E324A
Forward: 5′-CGGCTGAAACAGGCAGCACTGCTTCGGCAG-3′Reverse: 5′-CTGCCGAAGCAGTGCTGCCTGTTTCAGCCG-3′
R327A/Q328A
Forward: 5′-CAAGAACTGCTTGCGGCGGCAATGCGGAAT-3′Reverse: 5′-ATTCCGCATTGCCGCCGCAAGCAGTTCTTG-3′
R327Q/Q328R
Forward: 5′-CAAGAACTGCTTCAGCGGGCAATGCGGAAT-3′Reverse: 5′-ATTCCGCATTGCCCGCTGAAGCAGTTCTTG-3′
All constructs were confirmed by DNA sequencing (Eurofins Genomics).
Antibodies and reagents
The mouse monoclonal antibodies (mAbs) used in this study were anti-FLAG (M2) mAb (F-3165; Sigma-Aldrich, St. Louis, MO, USA), anti-HSC70 (B-6) mAb (sc-7298; Santa Cruz Biotechnology, Dallas, TX, USA), anti-c-Myc (9E10) mAb (sc-40; Santa Cruz Biotechnology), anti-ß-actin mAb (A-5441; Sigma-Aldrich), anti-DDDDK-tag mAb (M185-3L; MBL International Corporation, Woburn, MA, USA), anti-YAP1 (63.7) mAb (sc-101199; Santa Cruz Biotechnology), and anti-VPS4B (A-11) mAb (sc-377162; Santa Cruz Biotechnology). The rabbit polyclonal antibodies (pAbs) used in this study were anti-HA pAb (H-6908; Sigma-Aldrich), anti-YAP1 mAb (4912S; Cell Signaling Technology, Danvers, MA, USA), anti-Phospho-YAP1 (Ser127) mAb (4911S; Cell Signaling Technology), anti-LAMP2A pAb (ab18528; Abcam, Cambridge, UK), anti-DDDDK-tag pAb (PM020; MBL International Corporation) and anti-NS5A (2914-1) pAb (a kind gift from T. Wakita, National Institute of Infectious Diseases, Tokyo). Horseradish peroxidase (HRP)-conjugated anti-mouse IgG (7076S; Cell Signaling Technology) and HRP-conjugated anti-rabbit IgG (7074S; Cell Signaling Technology) were used as secondary antibodies.
Immunoblot analysis
Immunoblot analysis was performed as described previously [33, 34]. Cell lysates were resolved on 10% or 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to 0.45 μm polyvinylidene difluoride membrane (PVDF) (Immobilon-P; Millipore, Billerica, MA). Membranes were probed with primary antibodies, followed by horseradish peroxidase (HPR)-conjugated secondary antibody. The positive bands were visualized using Amersham enhanced chemiluminescence (ECL) western blotting detection reagents (RPN2106; Cytiva, Sigma-Aldrich). Band intensities were quantified using ImageJ software (version 1.53r).
Immunoprecipitation
Cells were lysed in buffer containing 150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 1% NP-40, 0.1% SDS, 1% sodium deoxycholic acid, 1 mM EDTA, and protease inhibitor cocktail (cOmplete™, EDTA-free, Roche Molecular Biochemicals, Mannheim, Germany). The lysate was centrifuged at 12,500 × g for 15 min at 4°C, and the supernatants were subjected to immunoprecipitation using appropriate antibodies. Immunoprecipitation was performed as described [35, 36]. Briefly, the cell lysates were immunoprecipitated with anti-FLAG M2 affinity gel (Sigma-Aldrich Co., A2220) or Protein A-Sepharose 4 Fast Flow (GE17-5280-04; GE Healthcare, Buckinghamshire, UK) pre-incubated with appropriate antibodies. Samples were rotated at 4°C for 4 h or overnight. After five washes with the lysis buffer, the immunoprecipitated samples were analyzed by immunoblotting.
RNA interference and generation of stable knockdown cells
The target sequences for the short hairpin RNAs (shRNAs) against LAMP2A, VPS4B, and scramble control were as follows:
shLAMP2A: 5′-GGCAGGAGUACUUAUUCUA-3′shVPS4B: 5′-AGCGAUAGAUCUGGCUAGCAA-3′shScramble: 5′-GGACAUCGACGGCUUUAUA-3′
A pair of complementary oligonucleotides encoding the shRNA targeting VPS4B was synthesized, annealed, and inserted into the pSilencer 2.1-U6 puro vector according to the manufacturer’s instructions (AM5762; Ambion, Austin, TX, USA). A pair of complementary oligonucleotides encoding the shRNA targeting LAMP2A was synthesized, annealed, and inserted into the pSilencer 2.1-U6 hygro vector (AM5760; Ambion). A pair of complementary oligonucleotides encoding the shRNA targeting scrambled control was inserted into the pSilencer 2.1-U6 hygro vector similarly. Huh-7.5 cells were transfected with these plasmids, and stable knockdown cell lines were established by selecting drug-resistant clones using 1 μg/ml puromycin (P9620; Sigma-Aldrich) or 200 μg/ml hygromycin B (09287-84; Nacalai Tesque, Kyoto, Japan) depending on the resistance cassette in the vector (6, 37).
Immunofluorescence staining
Huh-7.5 cells cultured on glass coverslips were fixed with 4% paraformaldehyde at room temperature (RT) for 15 min. After being washed with PBS, the cells were permeabilized with PBS containing 0.1% Triton X-100 for 15 min at RT. To block nonspecific binding, the cells were incubated in PBS supplemented with 1% bovine serum albumin (BSA) (01859-47; Nacalai Tesque) for 60 min. Subsequently, the cells were incubated for 60 min at RT with a mouse monoclonal anti-YAP1 antibody and a rabbit polyclonal anti-NS5A antibody, both diluted in 1% BSA in PBS. Afterwards, the cells were washed three times with PBS and further incubated for 60 min at RT with Alexa Fluor™ 594-conjugated anti-mouse IgG (A11005; Invitrogen) and Alexa Fluor™ 488-conjugated anti-rabbit IgG (A11008; Invitrogen) diluted in 1% BSA in PBS. Finally, the cells were washed four times with PBS, mounted on glass slides, and imaged using a confocal microscope (LSM 700, Carl Zeiss, Oberkochen, Germany).
Cell fractionation assay
Cytoplasmic and nuclear fractions were prepared from HCV-infected and mock-infected Huh-7.5 cells using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (78835; Thermo Fisher Scientific) according to the manufacturer’s instructions. The resulting lysates were subjected to immunoblot analysis.
RESULTS
The KFERQ motif at amino acid (aa) positions 324–328 of YAP1 is essential for interaction with HSC70
To identify potential KFERQ motifs in YAP1, we analyzed its aa sequence according to the consensus sequence rules for KFERQ motifs. This analysis revealed two possible motifs: one spanning aa residues 323–327 (QELLR), and another spanning aa residues 324–328 (ELLRQ) (Figs. 1A and 1B). Both sequences conform to the KFERQ consensus.
To assess the functional relevance of these motifs in mediating the interaction between YAP1 and HSC70, we generated point mutants of YAP1 expression plasmids: pCAG-HA-YAP1 Q323A, pCAG-HA-YAP1 Q323A/E324A, pCAG-HA-YAP1 R327A/Q328A, and pCAG-HA-YAP1 R327Q/Q328R (Fig. 1B). Co-immunoprecipitation analyses revealed that FLAG-HSC70 was co-precipitated with wild-type (WT) HA-YAP1, as well as with HA-YAP1 Q323A and HA-YAP1 Q323A/E324A (Fig. 1C, first panel, lanes 1, 2, and 3). In contrast, HA-YAP1 R327A/Q328A and HA-YAP1 R327Q/Q328R exhibited significantly reduced binding to FLAG-HSC70 (Fig. 1C, first panel, lane 4; Fig. 1D, first panel, lanes 7 and 8). These results indicate that the KFERQ motif at aa 324–328 (ELLRQ) is crucial for the YAP1-HSC70 interaction.
The substrate-binding domain of HSC70 mediates interaction with YAP1
To map the region of HSC70 responsible for binding to YAP1, we performed co-immunoprecipitation analysis using a series of FLAG-tagged HSC70 deletion mutants (Fig. 2B). HSC70 comprises two main domains: the N-terminal nucleotide-binding domain (NBD, aa 1–386), and the C-terminal substrate-binding domain (SBD, aa 394–646). The SBD is further divided into a 10-kDa helical LID domain (aa 509–604) and a 5-kDa dynamically unstructured C-terminal domain (CTD, aa 605–646). The HA-YAP1 proteins were coimmunoprecipitated with all of the FLAG-HSC70 proteins except FLAG-HSC70 (aa 1–386) (Fig. 2C, upper panel, lanes 3–6) with the use of anti-FLAG mAb. These results suggest that the SBD domain on HSC70 amino acid consisting of aa 394 to 509 is important for YAP1 binding. The SBD domain is critical for binding to the substrate protein for degradation. This result suggests that YAP1 is recognized by HSC70 as a substrate for selective degradation.
YAP1 is degraded via chaperone-mediated autophagy
To further investigate whether YAP1 is degraded via lysosomal or proteasomal degradation pathway, we treated the Huh-7.5 cells with the lysosomal inhibitor bafilomycin A1 or the proteasome inhibitor MG132. YAP1 expression increased upon bafilomycin A1 treatment but remained unchanged with MG132, suggesting lysosomal, rather than proteasomal, degradation (Figs. 3A and 3B, first panel). LAMP2A serves as a specific receptor for the CMA pathway, whereas VPS4B is essential for eMI. Therefore, to determine whether YAP1 is degraded via CMA or eMI, we generated stable LAMP2A knockdown Huh-7.5 cells (shLAMP2A-Huh-7.5 cells) and stable VPS4B knockdown Huh-7.5 cells (shVPS4B-Huh-7.5 cells) using short hairpin RNA (shRNA). Immunoblot analysis showed an increase in YAP1 levels in shLAMP2A-Huh-7.5 cells than in the shVPS4B-Huh-7.5 cells (Fig 3C, first panel). This finding suggests that YAP1 is degraded via CMA in Huh-7.5 cells.
HCV NS5A prevents the interaction between HSC70 and YAP1 protein
To determine whether HCV NS5A protein interacts with YAP1 protein in Huh-7.5 cells, we co-transfected pCAG-HA-YAP1 together with pCAG-FLAG-NS5A. Immunoprecipitation analysis using anti-FLAG mAb revealed that FLAG-HSC70 was co-precipitated with HA-YAP1 protein (Fig. 4A, first panel, lane 6), which is consistent with the results from Fig. 1C and Fig. 1D. However, FLAG-NS5A was not co-precipitated with HA-YAP1 (Fig. 4A, first panel, lane 5). These results suggest that HSC70 interacts with YAP1, whereas NS5A does not bind directly to YAP1 in Huh-7.5 cells.
Furthermore, YAP1 WT was co-precipitated with HSC70, but YAP1 S127A mutant co-precipitation was markedly reduced (Fig. 4B, first panel, lanes 8 and 9). Serine 127 phosphorylation is known to facilitate YAP1 retention in the cytoplasm. Therefore, lack of phosphorylation at this site correlates with increased YAP1 translocation into the nucleus. This result suggests that HSC70 primarily associates with cytoplasmic YAP1.
To investigate a possible role of NS5A in the interaction between HSC70 and YAP1, Huh-7.5 cells were transfected with increasing amounts of either pEF1A-NS5A-Myc-His_6_ or pEF1A-NS4B-Myc-His_6_. Overexpression of NS5A-Myc-His_6_ (Fig. 4C, upper panels, lane 4), but not NS4B-Myc-His_6_ (Fig. 4D, upper panels, lane 4), resulted in significantly reduced co-immunoprecipitation of HA-YAP1 with FLAG-HSC70. These findings suggest that NS5A disrupts interaction between HSC70 and YAP1.
HCV infection increases YAP1 protein levels via NS5A
To investigate the effect of HCV infection on YAP1 protein levels, we used Huh-7 cells stably harboring the HCV-1b full genome replicon (RCYM1). Immunoblot analysis revealed that YAP1 protein levels were elevated in HCV-infected cells (RCYM1) compared to control Huh-7 cells (Fig. 5A, first panel, lanes 2 and 4). Consistent with this, increased YAP1 levels were also observed in Huh-7.5 cells infected with HCV J6/JFH1 (Fig. 5B, first panel, lane 2).
To examine whether NS5A contributes to the HCV-induced YAP1 stabilization, Huh-7.5 cells were transfected with increasing amounts of either pCAG-FLAG-NS5A, pCAG-FLAG-Core, or pEF1A-NS4B-Myc-His_6_. NS5A overexpression, but not core or NS4B protein, increased YAP1 protein levels (Fig. 5C, first panel, lanes 2 and 3). These findings indicate that HCV infection promotes YAP1 protein stabilization.
HCV infection induces YAP1 nuclear translocation
To further investigate the effect of HCV on YAP1, we performed immunofluorescence staining to examine YAP1 subcellular localization in mock and HCV-infected Huh-7.5 cells. In the mock-infected group, YAP1 was distributed in both the nucleus and cytoplasm, as shown in the fluorescence image and the corresponding line scan profile (Figs. 6A and 6B). In contrast, YAP1 was predominantly localized in the nucleus in HCV-infected cells (Fig. 6A). This is supported by the line scan profile, in which the YAP1 fluorescence intensity peak overlaps with the Hoechst33342 nuclear signal peak, indicating YAP1 accumulation within the nucleus (Fig. 6C).
To biochemically confirm these findings, we performed cell fractionation assay. Consistent with the immunofluorescence staining, the cell fractionation assay showed that HCV infection led to a decrease in cytoplasmic YAP1 (Fig. 6B) and an increase in nuclear YAP1 (Fig. 6C). These findings indicate that HCV infection promotes YAP1 nuclear translocation.
DISCUSSION
We previously demonstrated that the HCV NS5A protein interacts with the cellular chaperone protein HSC70 during HCV infection. The interaction between NS5A and HSC70 promotes the binding of HSC70 to the KFERQ motif on target proteins, and induces the lysosomal degradation of proteins, such as HNF-1α degradation via CMA and DGAT1 degradation via eMI. The degradation of these host factors contributes to HCV pathogenesis by altering cellular environment in a way that supports efficient viral replication [2–6]. In this study, we focused on the transcriptional co-activator YAP1 protein that plays a crucial role in cell proliferation. We identified possible KFERQ motifs located at aa 323–327 and 324–328 within YAP1 and examined its interaction with HSC70 by generating YAP1 mutants lacking key residues within the possible motifs. Notably, the R327A/Q328A mutation significantly reduced the binding to HSC70, indicating that the ^324^ELLRQ^328^ sequence functions as a bona fide KFERQ motif recognized by HSC70. HSC70 comprises two major domains: NBD and SBD. Using deletion mutants of HSC70, we demonstrated that the SBD is essential for binding to YAP1. Furthermore, treatment with a lysosomal inhibitor, but not a proteasomal inhibitor, led to stabilization of YAP1. These findings indicate that YAP1 is targeted for degradation via selective lysosomal degradation pathway in HSC70-dependent manner.
We next examined whether this HSC70-mediated degradation of YAP1 occurs through CMA or eMI. Since different receptors are involved in each pathway, we knocked down LAMP2A for CMA and VPS4B for eMI to distinguish two pathways. In LAMP2A knockdown cells, YAP1 levels increased, suggesting that YAP1 is degraded via CMA. In contrast, the knockdown of VPS4B had little effect on YAP1 levels, suggesting that eMI is not involved in degradation of YAP1.
To investigate the effect of NS5A on the HSC70–YAP1 interaction, we transfected cells with increasing amounts of NS5A. While NS5A did not directly interact with YAP1, NS5A inhibited HSC70-YAP1 binding in a dose-dependent manner. HCV infection led to increased YAP1 protein levels and enhanced nuclear translocation of YAP1. Importantly, overexpression of NS5A, but not other HCV proteins such as NS4B or Core, led to elevated YAP1 protein levels. We propose a model in which under normal condition, HSC70 interacts with YAP1 to mediate YAP1 degradation via CMA. During HCV infection, NS5A binds to HSC70 and redirects HSC70 to NS5A-binding proteins, thereby preventing HSC70 from targeting YAP1 for degradation. As a result, YAP1 accumulates and translocates into the nucleus (Fig. 7).
We also observed that HA-YAP1 S127A lacking phosphorylation at serine 127 showed reduced binding to HSC70. This suggests that serine 127 phosphorylation is important for YAP1–HSC70 interaction. It is also possible that the reduced HSC70-binding to YAP1 S127A mutant is due to its decreased cytoplasmic localization, as phosphorylation at serine 127 is known to promote cytoplasmic retention of YAP1 [38], where HSC70 can access its targets. Further analysis is required to determine the detailed mechanism.
In addition to our proposed model, in which NS5A directly interacts with HSC70 to disrupt its binding to phosphorylated YAP1, alternative mechanisms may also account for our observations. One possibility is that NS5A modulates upstream components of the Hippo pathway, such as by inhibiting LATS1/2 kinase activity or by activating an unidentified phosphatase, resulting in an increase in non-phosphorylated YAP1. In this study, we demonstrated that HSC70 preferentially binds to YAP1 phosphorylated at serine 127. Therefore, a reduction in YAP1 phosphorylation at this site could weaken its interaction with HSC70, allowing more YAP1 to escape chaperone-mediated autophagy and translocate into the nucleus. While our current findings support a model involving direct NS5A-HSC70 interaction, further studies are required to determine whether NS5A also affects upstream Hippo pathway regulators or phosphatase activity.
Phosphorylation may also broadly influence CMA substrate specificity. For example, certain proteins, such as PLIN2, require phosphorylation to become CMA substrates, whereas others, like phosphoprotein enriched in diabetes (PED), are degraded only in their unphosphorylated state [39–42]. It has been proposed that phosphorylation of serine, threonine, or tyrosine residues near an incomplete KFERQ-like motif can introduce a negative charge, thereby converting it into a functional CMA-targeting signal [10]. However, YAP1 already contains a canonical KFERQ motif, and Ser127 is not located within this motif, suggesting an alternative mechanism–perhaps a phosphorylation-induced conformational change that enhances HSC70 accessibility. Similar regulatory mechanisms have been described for hypoxia-inducible factor 1-alpha (HIF1α), which requires ubiquitination, and mammalian Ste 20-like kinase 1 (MST1), which becomes accessible to CMA only after deacetylation [43, 44].
In conclusion, our findings reveal a novel mechanism by which NS5A disrupts the HSC70-YAP1 interaction, thereby stabilizing YAP1 by inhibiting its CMA-mediated degradation and promoting nuclear translocate of YAP1. Nuclear YAP1 may subsequently activate target genes involved in cell proliferation, potentially creating a favorable environment for viral replication. Further studies are warranted to clarify the mechanism of HCV-induced YAP1 nuclear translocation and to explore its broader implications in viral pathogenesis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Krause GJ Kirchner P Stiller B Morozova K Diaz A Chen KH Molecular determinants of the crosstalk between endosomal microautophagy and chaperone-mediated autophagy Cell Rep 202342121135293806038010.1016/j.celrep.2023.113529 PMC 10807933 · doi ↗ · pubmed ↗
- 2Matsui C Deng L Minami N Abe T Koike K Shoji I Hepatitis C Virus NS 5A Protein Promotes the Lysosomal Degradation of Hepatocyte Nuclear Factor 1α via Chaperone-Mediated Autophagy Pfeiffer JKJ Virol 20189213 e 00639182969541910.1128/JVI.00639-18PMC 6002715 · doi ↗ · pubmed ↗
- 3Matsui C Rosalyn Sianipar I Minami N Deng L Hotta H Shoji IA single-amino-acid mutation in hepatitis C virus NS 5A disrupts physical and functional interaction with the transcription factor HNF-1αJ Gen Virol 2015968220052595709710.1099/vir.0.000179 · doi ↗ · pubmed ↗
- 4Matsui C Shoji I Kaneda S Sianipar IR Deng L Hotta H Hepatitis C Virus Infection Suppresses GLUT 2 Gene Expression via Downregulation of Hepatocyte Nuclear Factor 1αJ Virol 2012862312903112299315010.1128/JVI.01418-12PMC 3497665 · doi ↗ · pubmed ↗
- 5Matsui C Yuliandari P Deng L Abe T Shoji I The Role of Chaperone-Mediated Autophagy in Hepatitis C Virus-Induced Pathogenesis Front Cell Infect Microbiol 2021117966643492633010.3389/fcimb.2021.796664 PMC 8674663 · doi ↗ · pubmed ↗
- 6Yuliandari P Matsui C Deng L Abe T Mori H Taguwa S Hepatitis C virus NS 5A protein promotes the lysosomal degradation of diacylglycerol O-acyltransferase 1 (DGAT 1) via endosomal microautophagy Autophagy Rep 202211264854039603510.1080/27694127.2022.2095591 PMC 11864691 · doi ↗ · pubmed ↗
- 7Evans CG Chang L Gestwicki JE Heat Shock Protein 70 (Hsp 70) as an Emerging Drug Target J Med Chem 2010531245856022033436410.1021/jm 100054 f PMC 2895966 · doi ↗ · pubmed ↗
- 8Kaushik S Cuervo AM Chaperone-mediated autophagy: a unique way to enter the lysosome world Trends Cell Biol 2012228407172274820610.1016/j.tcb.2012.05.006PMC 3408550 · doi ↗ · pubmed ↗