BEREN: a bioinformatic tool for recovering giant viruses, polinton-like viruses, and virophages in metagenomic data
Benjamin Minch, Mohammad Moniruzzaman

TL;DR
BEREN is a new bioinformatic tool designed to recover and analyze giant viruses and related viruses in metagenomic data.
Contribution
The novel contribution is BEREN, a comprehensive tool specifically optimized for recovering NCLDV and Preplasmiviricota viruses from metagenomes.
Findings
BEREN outperformed existing tools in recovering NCLDV contigs and Preplasmiviricota genomes from a mock metagenome.
The tool includes modules for genome recovery, marker gene detection, and metabolic protein annotation.
BEREN provides a user-friendly solution for studying the ecological roles of eukaryotic viruses.
Abstract
Viruses in the kingdom Bamfordvirae, specifically giant viruses (NCLDVs) in the phylum Nucleocytoviricota and smaller members in the Preplasmiviricota phylum, are widespread and important groups of viruses that infect eukaryotes. While viruses in this kingdom, such as giant viruses, polinton-like viruses, and virophages, have gained large interest from researchers in recent years, there is still a lack of streamlined tools for the recovery of their genomes from metagenomic datasets. Here, we present, BEREN, a comprehensive bioinformatic tool to unlock the diversity of these viruses in metagenomes through five modules for NCLDV genome, contig, and marker gene recovery, metabolic protein annotation, and Preplasmiviricota genome identification and annotation. BEREN’s performance was benchmarked against other mainstream virus recovery tools using a mock metagenome, demonstrating superior…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
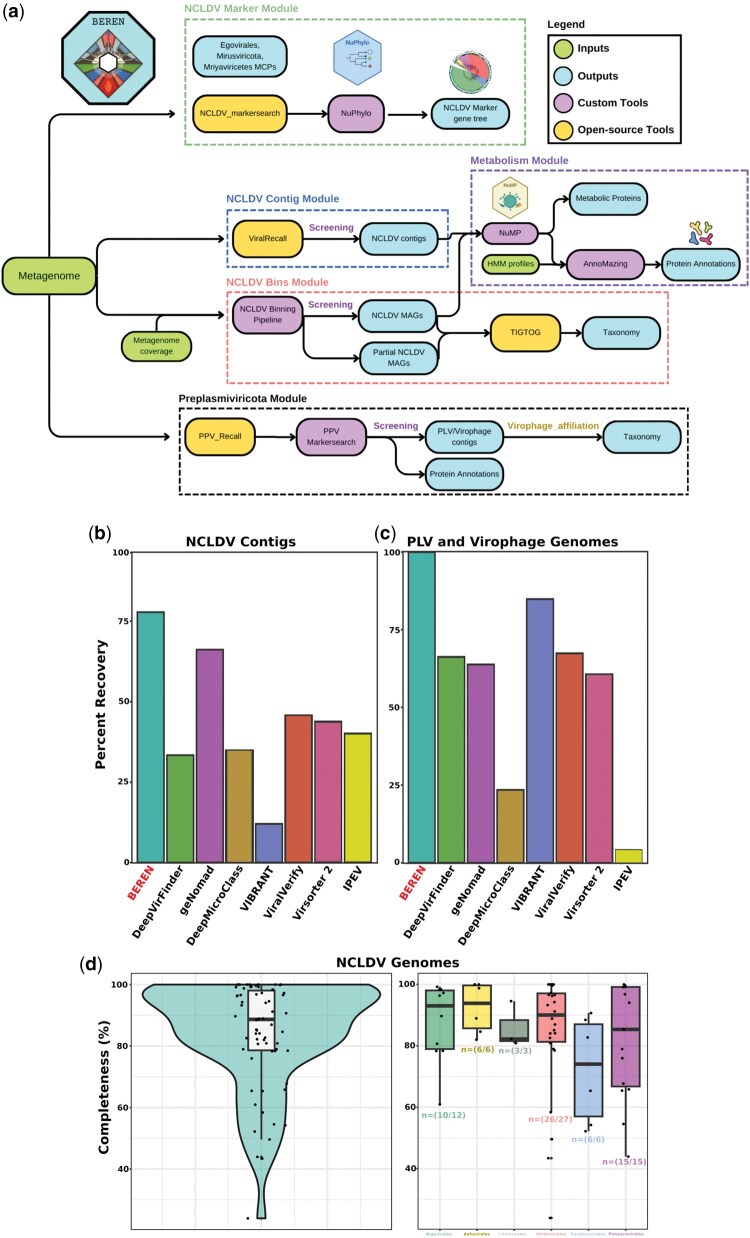 Figure 1
Figure 1- —Rosenstiel School of Marine, Atmospheric, and Earth Sciences, University of Miami to Dr. Mohammad Moniruzzaman
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacteriophages and microbial interactions · Plant Virus Research Studies · Genomics and Phylogenetic Studies
1 Introduction
The Eukaryotic viruses are widespread and important members of the global virosphere, infecting both charismatic macrofauna such as pigs and smaller microeukaryotes like photosynthetic algae. One of the major kingdoms of these viruses is Bamfordvirae, which includes the Nucleocytoviricota phylum (giant viruses) (Aylward et al. 2021) and the closely related Preplasmiviricota phylum, which contains virophages, polintons, and polinton-like viruses (Koonin et al. 2024). Recent research has highlighted the widespread presence of viruses from these two phyla of double-stranded DNA viruses in diverse ecosystems, including both terrestrial and aquatic environments (Bellas and Sommaruga 2021; Minch et al. 2023; Schulz et al. 2018, 2020). These viruses also often infect environmentally important microeukaryotic hosts such as the bloom-forming algae Emiliania huxleyi and Phaeocystis globosa (Bratbak et al. 1993; Juneau et al. 2003; Vincent et al. 2021).
While many bioinformatic tools exist for recovering viruses from metagenomes, most of these tools have a strong emphasis on prokaryotic viruses such as phages infecting bacteria (Wu et al. 2024). Some recent tools, such as geNomad and IPEV (Camargo et al. 2024; Yin et al. 2024), can identify dsDNA eukaryotic viruses, but these tools often lack specificity as to the identity of the eukaryotic virus or the ability to recover full genomes of these viruses for comprehensive downstream analysis. The recovery of full genomes is especially of concern for giant viruses, as their genomes are often too large to be assembled into a single contig through short-read sequencing, and no current tool is fit to recover full genomes. In addition, these tools do not take into account the ever-expanding diversity of these viruses, such as the newly discovered Mirusviricota phylum (Gaïa et al. 2023), Egovirales order (Gaïa et al., 2024), or Mriyaviricetes class (Yutin et al. 2024).
There is currently no consensus methodology or comprehensive tool for recovery of these viruses (Bellas and Sommaruga 2021; Moniruzzaman et al. 2020; Pitot et al. 2024; Schulz et al. 2020), and many of the proposed methods require extensive bioinformatic expertise and manual screening. Here we present BEREN (Bioinformatic tool for Eukaryotic virus Recovery from Environmental metagenomes), a “one-stop-shop” for uncovering the diversity and metabolic potential of giant viruses, polinton-like viruses, and virophages in any metagenomic sample. This tool both opens up the realm of Bamfordvirae viruses to interested researchers and provides a streamlined methodology to increase repeatability. We demonstrate that this tool outperforms available virus recovery tools in recovering NCLDV contigs and Preplasmiviricota genomes, as well as provides extra functionality integrated in the pipeline, such as genome binning, taxonomy, decontamination, and metabolic annotation.
2 Methods
2.1 The BEREN tool
The BEREN tool comprises five different modules that can be run independently or all together on a metagenomic dataset. These modules include (1) NCLDV markers, (2) NCLDV contigs, (3) NCLDV bins, (4) metabolism and protein annotation, and (5) Preplasmiviricota identification. The tool is publicly available at: https://gitlab.com/benminch1/BEREN. Detailed instructions and necessary scripts for installation and database downloads are also provided there, along with default parameters for the tools integrated in the BEREN workflow (Fig. 1).
The BEREN pipeline and benchmarking. (a) BEREN consists of 5 major modules, each comprising multiple open-source tools as well as custom scripts. The input of a metagenome can yield NCLDV markers, contigs, and genomic bins, Preplasmiviricota genomes, as well as the metabolic potential and protein annotation of these viruses. The recovery of (b) NCLDV contigs and (c) PLV and virophage genomes was benchmarked against popular virus identification tools. Each tool was run with default settings, and total recovery is shown here. (d) The completeness of the recovered NCLDV genomes from BEREN was tested by comparing recovered genomes to those used to build the mock metagenome. Completeness is defined as the percentage of the total length of the genome recovered. This information is also provided for each NCLDV order, with the number of recovered genomes in that order below the boxplots.
2.1.1 NCLDV marker module
The main purpose of the NCLDV marker module is to gain insight into the full diversity of NCLDVs within a given metagenomic dataset, including those that remain elusive to genome binning methods due to low coverage or abundance, as well as newly discovered NCLDV relatives such as Egoviruses, Mirusviruses, and Mriyaviruses. The NCLDV marker module first predicts proteins from the metagenomic assembly using prodigal-gv (Camargo et al. 2024; Hyatt et al. 2010). It then searches for NCLDV marker genes with the NCLDV_markersearch script (Aylward et al. 2021). The marker genes searched for include the NCLDV major capsid protein (MCP), DEAD/SNF2-like helicase (SFII), DNA-directed RNA polymerase beta and alpha subunits (RNAPS and RNAPL), DNA polymerase family B (PolB), Transcription initiation factor IIB (TFIIB), DNA topoisomerase II (TopoII), Packaging ATPase (A32), and the Poxvirus Late Transcription Factor VLTF3 (VLTF3). These markers were chosen based on their near universality in NCLDV genomes and their use in phylogenetic placement of these viruses (Aylward et al. 2021).
After storing all markers in separate protein FASTA files, this module will use the newly discovered PolB markers in the target metagenome dataset to build a phylogenetic tree (see Supplementary Methods). The sequences are first aligned using MAFFT (Katoh et al. 2002) to a custom set of reference NCLDV PolBs representing all NCLDV families, as well as a set of PolBs from bacteria and eukaryotes for quality filtering. The alignment is then trimmed with trimAL using the “-gt 0.1” parameter (Capella-Gutiérrez et al. 2009), and a phylogenetic tree is constructed using fasttree (Price et al. 2010).
2.1.2 NCLDV contig module
While NCLDV genome recovery is an obvious goal of many researchers wanting to study these viruses, recovery of full genomes might not always be possible due to fragmented assemblies and known chimerism of NCLDVs (Moreira and Brochier-Armanet 2008; Yu et al. 2024). In the NCLDV contig pipeline, NCLDV contigs are retrieved using ViralRecall (Aylward and Moniruzzaman 2021), which identifies NCLDV contigs based on homology to known NCLDV proteins. The identified contigs are screened for an NCLDV score chosen by the user (default of 3) and a length of >10kbp. The method for deriving these scores is described in Aylward and Moniruzzaman (2021).
2.1.3 NCLDV bins module
This module is designed to recover both high-quality and partial NCLDV genomes through a genome binning, screening, and cleaning pipeline. First, metagenomic contigs are binned using metabat2 (Kang et al. 2019), and then potential NCLDV bins are identified using the NCLDV markersearch script (Aylward et al. 2021) after proteins are predicted using prodigal-gv (Camargo et al. 2024). Bins with ≥1 hit to an NCLDV marker gene are retained for further analysis. These bins are then screened using ViralRecall in “contig” mode (Aylward and Moniruzzaman 2021) to confirm they are NCLDVs. Bins with negative ViralRecall scores (those with more hits to cellular protein groups than viral ones) are further investigated to assess whether they represent partial NCLDV genomes.
After this screening, another screening is performed to further separate “high-quality” and “partial” NCLDV bins. High-quality NCLDV bins are defined as having at least 3 of the following marker genes (PolB, MCP, A32, VLTF3, SFII) so they can be easily used in a concatenated phylogeny (Aylward et al. 2021). These markers were chosen due to their highly conserved nature in NCLDVs (Aylward et al. 2021; Moniruzzaman et al. 2020). Both partial and high-quality bins are then processed using ViralRecall in “contig” mode, which screens individual contigs within the bins to remove potential cellular contamination. Taxonomy for these bins is assigned using TIGTOG, a tool that predicts taxonomy through a machine learning approach (Ha and Aylward 2024).
The NCLDV bins module can also potentially recover Mirusvirus and Mryiavirus genomes. The supplementary text discusses this in more detail (Fig. S1).
2.1.4 Metabolism and protein annotation module
Many NCLDVs encode genes for a wide range of cellular metabolic processes, potentially enabling them to reprogram cellular metabolism in myriad different ways, which is unprecedented in the virus world (Minch and Moniruzzaman 2025; Moniruzzaman et al. 2023). The metabolism and protein annotation module attempts to uncover these unique functions of NCLDVs through HMM-based protein annotation using multiple databases. Using recovered NCLDV contigs, NCLDV high-quality bins, and partial bins, this module first annotates proteins using Pfam (Mistry et al. 2021), GVOG (Aylward et al. 2021), and KEGG (Kanehisa et al. 2017) databases. Metabolic proteins of interest are parsed into a separate file using a custom script (github.com/BenMinch/nump). Resulting protein annotations can be used to analyze the functional profiles of the whole NCLDV community or individual NCLDV genomes.
2.1.5 Preplasmiviricota module
Virophages and Polinton-like viruses (PLVs) are all part of Preplasmiviricota, a sister phylum to NCLDVs (Koonin et al. 2024). These smaller eukaryotic viruses are widespread in aquatic ecosystems (Bellas and Sommaruga 2021) and often interact with NCLDVs (Roux et al. 2017), making their recovery important for understanding eukaryotic virus ecology. The Preplasmiviricota module effectively recovers and identifies these two types of viruses (Virophages and PLVs) in the Preplasmiviricota phylum in metagenomic datasets (see supplemental methods).
First, contigs potentially belonging to viruses in this phylum are screened using a modified version of ViralRecall with new protein profiles common across diverse virophage and PLV members (Stephens et al. 2024). Contigs with a positive score are then screened to only keep contigs with a major capsid protein as well as one other marker gene (integrase, minor capsid protein, and Ftsk ATPase). Taxonomy is assigned to these contigs using the Virophage_affiliation script (Roux et al. 2017). Protein annotations for these contigs are performed using both the Pfam database (Mistry et al. 2021) and a custom HMM profile of environmental virophage and PLV protein clusters (Bellas and Sommaruga 2021).
2.2 Benchmarking BEREN
2.2.1 Mock metagenome creation
To test the capabilities of the BEREN tool at recovering diverse NCLDV and Preplasmiviricota members, a mock metagenome was created using CAMISIM (Fritz et al. 2019) [size = 3, 150 bp mean length]. To create the mock community, 100 genomes of unique virophages and PLVs (Bellas and Sommaruga 2021) were obtained (dereplicated at 80% ANI using dRep (Olm et al. 2017)) as well as 69 unique NCLDV genomes from the GOEV database (Gaïa et al. 2023)(637 total contigs), representing 3 from each major NCLDV family, covering all NCLDV orders. Ten bacterial genomes were also used to simulate ubiquitous bacterial populations within environmental metagenomic samples, and these genomes were obtained from the GORG tropics database (Pachiadaki et al. 2019).
2.2.2 Benchmarking BEREN and other virus recovery tools
The BEREN tool was benchmarked against a set of other popular virus recovery tools, including DeepVirFinder (Ren et al. 2020), geNomad (Camargo et al. 2024), DeepMicroClass (Hou et al. 2024), VIBRANT (Kieft et al. 2020), ViralVerify (Antipov et al. 2020), Virsorter2 (Guo et al. 2021), and IPEV (Yin et al. 2024). While most of these tools were built with prokaryotic viruses in mind, they can often perform moderately well at recovering eukaryotic viruses (Camargo et al. 2024).
All tools were run with default settings using the mock metagenome as input. Many of these tools (with the exception of geNomad) will not explicitly discriminate between NCLDV and Preplasmiviricota viruses, so sequences identified as viral were run through the BEREN pipeline to parse these two groups apart. For tools using deep learning (DeepVirFinder, DeepMicroClass, and IPEV), a score cutoff of 0.9 was used to determine viral sequences. The percent recovery of NCLDV contigs, as well as Preplasmiviricota genomes, was evaluated for each tool.
2.2.3 Benchmarking BEREN for NCLDV genome completeness
To assess the ability of BEREN to recover high-quality “complete” NCLDV genomes, the NCLDV bin module was run on the mock metagenome. Each recovered genome bin was compared to the known genomes in the mock community using BLASTn (Camacho et al. 2009). Completeness was defined to be the percentage of the total genome length recovered by BEREN.
3 Results
3.1 BEREN exceeds other automated virus detection tools in NCLDV and preplasmiviricota recovery
A benchmarking of the BEREN tool against other popular viral recovery tools (DeepVirFinder, geNomad, DeepMicroClass, VIBRANT, ViralVerify, Virsorter2, and IPEV) showed BEREN outperforms these tools on both NCLDV contig and Preplasmiviricota genome retrieval from the mock metagenome. Out of the 637 NCLDV contigs above 10 kbp in the mock metagenome, BEREN was able to recover 489 contigs (77%) (Fig. 1b). The next best tool, geNomad, was able to identify 63% of these contigs, with many tools recovering below 50%. It is important to note that these contigs do not represent full NCLDV genomes, as many genomes contain multiple contigs.
Recovery of Preplasmiviricota genomes gave similar results as BEREN recovered all (100%) of the 100 genomes seeded into the mock metagenome (Fig. 1c). Other tools recovered between 4% and 83% of the Preplasmiviricota genomes.
3.2 BEREN recovers high-quality NCLDV genomes
Out of the 69 seeded NCLDV genomes, BEREN was able to recover 66 (95.6%). Using BLASTn to match the recovered genomes with the seeded genomes, we were able to calculate the completeness of each recovered genome. Genomes recovered with BEREN had an average completeness of 84% and a median completeness of 89% (Fig. 1d).
3.3 Testing BEREN with real-world metagenomes
As a demonstration of its potential use, BEREN was run on a metagenomic dataset from the Baltic Sea. The results from this analysis can be found in the supplementary text (Fig. S2).
4 Discussion
In addition to outperforming other viral identification tools in the realm of NCLDVs and Preplasmiviricota recovery, BEREN has several other advantages. One of such advantages is that BEREN is designed specifically for these two groups of viruses and, therefore, provides many helpful downstream outputs for studying their diversity, such as marker genes for phylogenetic and taxonomic assignment. While several other tools were able to recover NCLDVs and Preplasmiviricota viruses, many of them do not provide any distinguishing taxonomic or phylogenetic information about these viruses. For example, DeepMicroClass and IPEV label both these phyla as “Eukaryote Virus,” and other tools like VIBRANT and DeepVirFinder do not distinguish them from bacterial viruses.
Another advantage of BEREN is the ability to recover NCLDV genomic bins in addition to contigs. As far as we know, no other virus-identification tool tested can recover NCLDV genomic bins, which is a key limitation, as NCLDV genomes are often too large to be represented by a single contig (Aylward et al. 2021). Getting genomic bins also has the advantage of providing genome annotations to uncover the functional potential of these viruses.
Supplementary Material
vbaf284_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Antipov D , Raiko M, Lapidus A et al Metaviral SP Ades: assembly of viruses from metagenomic data. Bioinformatics 2020;36:4126–9. 10.1093/bioinformatics/btaa 49032413137 · doi ↗ · pubmed ↗
- 2Aylward FO , Moniruzzaman M, Ha AD et al A phylogenomic framework for charting the diversity and evolution of giant viruses. P Lo S Biol. 2021;19:e 3001430. 10.1371/journal.pbio.300143034705818 PMC 8575486 · doi ↗ · pubmed ↗
- 3Aylward FO , Moniruzzaman M. Viral Recall—a flexible command-line tool for the detection of giant virus signatures in ‘omic data. Viruses 2021;13:150. 10.3390/v 1302015033498458 PMC 7909515 · doi ↗ · pubmed ↗
- 4Bellas CM , Sommaruga R. Polinton-like viruses are abundant in aquatic ecosystems. Microbiome 2021;9:13. 10.1186/s 40168-020-00956-033436089 PMC 7805220 · doi ↗ · pubmed ↗
- 5Bratbak G , Egge J, Heldal M. Viral mortality of the marine alga Emiliania huxleyi (haptophyceae) and termination of algal blooms. Mar Ecol Prog Ser 1993;93:39–48. 10.3354/meps 093039 · doi ↗
- 6Camacho C , Coulouris G, Avagyan V et al BLAST+: architecture and applications. BMC Bioinformatics 2009;10:421. 10.1186/1471-2105-10-42120003500 PMC 2803857 · doi ↗ · pubmed ↗
- 7Camargo AP , Roux S, Schulz F et al Identification of mobile genetic elements with ge Nomad. Nat Biotechnol 2024;42:1303–12. 10.1038/s 41587-023-01953-y 37735266 PMC 11324519 · doi ↗ · pubmed ↗
- 8Capella-Gutiérrez S , Silla-Martínez JM, Gabaldón T. trim Al: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009;25:1972–3. 10.1093/bioinformatics/btp 34819505945 PMC 2712344 · doi ↗ · pubmed ↗