A PBD-dimer containing antibody drug conjugate targeting CCRL2 for high-risk MDS/AML
Theodoros Karantanos, Nour Naji, Taha Ahmedna, J Peske, Xinghan Zeng, Brandy Perkins, Zanshé Thompson, Tushar Nichakawade, Bum Lee, Evangeline Watson, Theodora Chatzilygeroudi, Li Luo, Bogdan Paun, Melanie Klausner, Yuju An, Teodora Supeanu, Ivana Gojo, Gabriel Ghiaur

TL;DR
A new antibody-drug conjugate targeting CCRL2 shows strong effectiveness against high-risk MDS/AML, especially in TP53-mutated cases.
Contribution
Development of a novel anti-CCRL2 ADC with superior cytotoxicity and efficacy in TP53-mutated MDS/AML models.
Findings
The anti-CCRL2 ADC showed stronger cytotoxicity than existing ADCs in TP53-mutated MDS/AML cell lines.
The ADC suppressed leukemic growth in xenograft models without harming healthy hematopoietic cells.
TP53-mutated MDS/AML and AML with erythroid features exhibited the highest CCRL2 expression.
Abstract
Patients with myelodysplastic syndrome (MDS)/acute myeloid leukemia (AML) with high-risk features including TP53 mutations have poor outcomes due to lack of effective therapies. The atypical chemokine surface receptor C-C motif chemokine receptor-like 2 (CCRL2) is overexpressed in MDS and secondary AML (sAML) compared to healthy hematopoietic cells and we recently found that TP53-mutated MDS/AML and AML with erythroid features express the highest levels of this receptor across MDS/AML subtypes. To illustrate the therapeutic potential of CCRL2 as a therapeutic target, we developed an anti-CCRL2 antibody-drug conjugate (ADC) by conjugating an anti-CCRL2 antibody with the cytotoxic drug pyrrolobenzodiazepine (PBD), which causes DNA double-strand breaks leading to cancer cell death. The anti-CCRL2 ADC demonstrated strong CCRL2-selective cytotoxicity against cell lines derived from MDS/AML…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
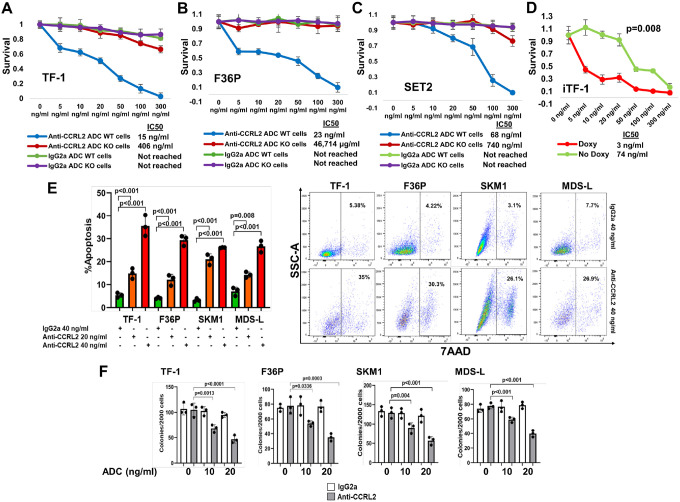 Figure 1
Figure 1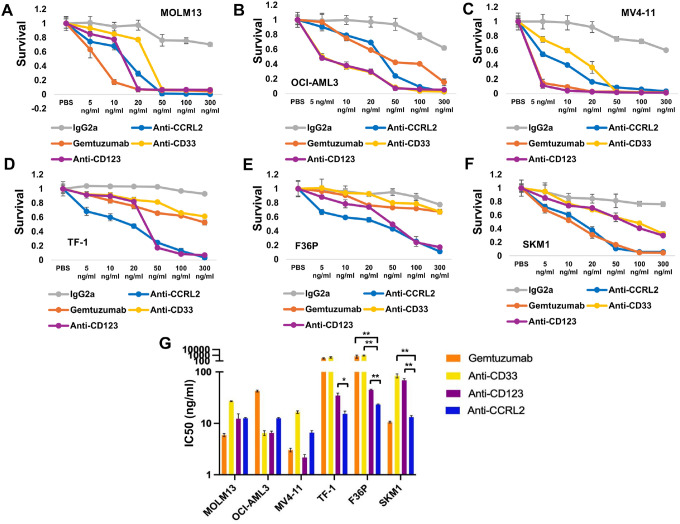 Figure 2
Figure 2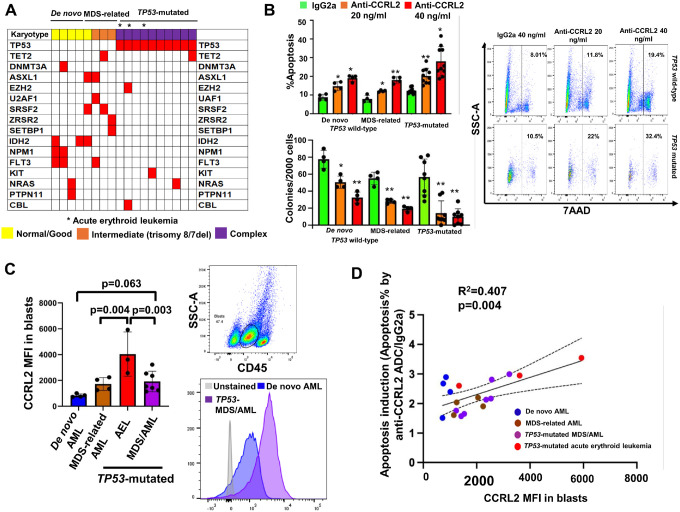 Figure 3
Figure 3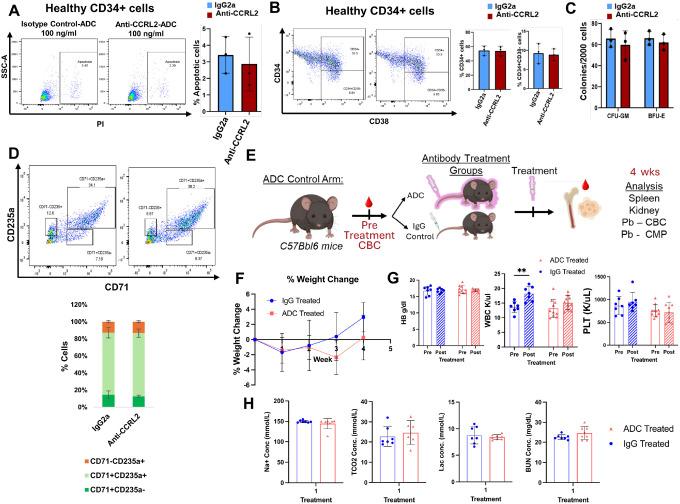 Figure 4
Figure 4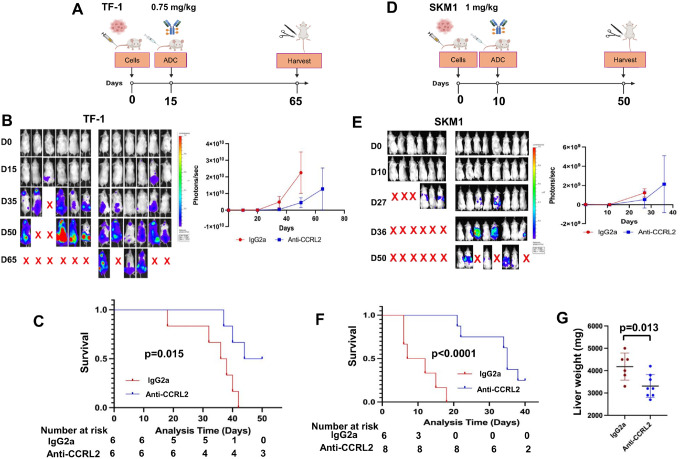 Figure 5
Figure 5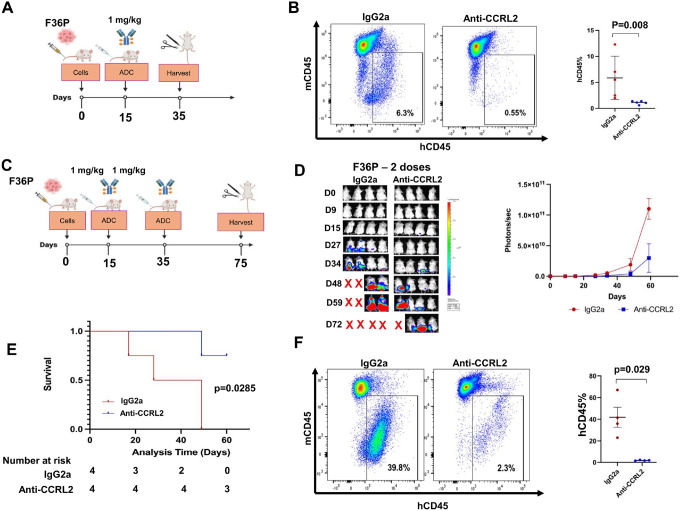 Figure 6
Figure 6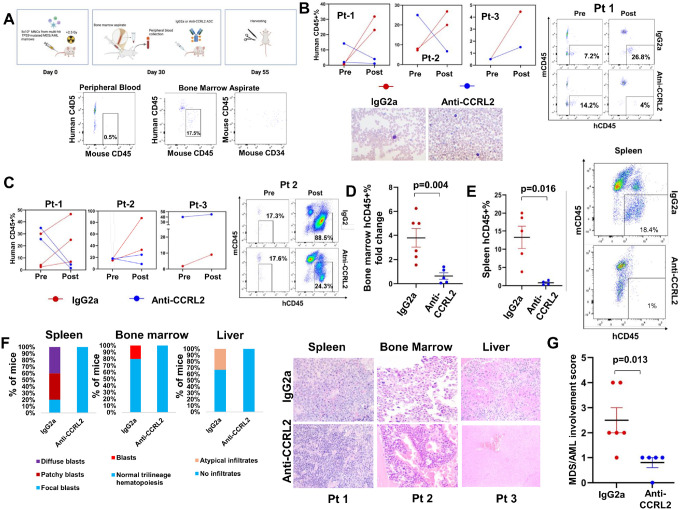 Figure 7
Figure 7Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAcute Myeloid Leukemia Research · Chemokine receptors and signaling · Multiple Myeloma Research and Treatments
Introduction
Patients with high-risk myelodysplastic syndrome (MDS) and secondary acute myeloid leukemia (sAML) continue to exhibit poor outcomes, especially in the presence of high-risk features such as TP53 mutations or deletions (1–3) Thus, the development of effective therapies is urgently needed for these individuals. Antibody-drug conjugates (ADCs) (4, 5), show prominent activity in lymphoid cancers(6, 7), and good-risk AML (8, 9). However, they demonstrate limited efficacy in high-risk MDS/AML(10).
The atypical chemokine receptor C-C motif chemokine-like receptor 2 (CCRL2) is normally expressed in differentiated myeloid cells contributing to the regulation of cells migration in inflammation cites (11–13). This surface receptor is upregulated in MDS and sAML, while normal hematopoietic progenitors demonstrate minimal expression(14). CCRL2 silencing suppresses MDS/sAML cell growth and sensitizes them to hypomethylating agents but does not affect the survival and clonogenicity of healthy CD34 + cells(14, 15). We recently discovered that across MDS/AML subtypes, TP53 mutated MDS/AML and AML with erythroid features express the highest CCRL2 levels. Intriguingly, TP53 deletion in AML TP53 wild-type (WT) cells caused a direct prominent CCRL2 upregulation(16). The main downstream target of CCRL2 is interferon gamma signaling, which has been associated with AML clonal evolution, TP53 deletion, acquisition of erythroid features and treatment resistance(16–18). Thus, CCRL2 is potentially a promising target in in high-risk MDS/sAML.
In this study, we show that an ADC targeting CCRL2 exhibits significant anti-leukemic effects in high-risk MDS/AML, including TP53-mutated cells, but has limited toxicity against normal hematopoietic cells.
Material and Methods
Cell lines and reagents
TF-1, SKM1, SET2, and MV4–11 cell lines were purchased from the American Type Culture Collection. F36P, OCI-AML3 and MOLM13 cells were purchased from Leibniz Institute DSMZ. MDS-L cells were a gift from Dr. Starczynowski, University of Cincinnati(19). MV4–11 cells were cultured in IMDM (Thermo Fischer Scientific, Waltham, MA) with 10% fetal bovine serum (FBS) (MilliporeSigma, Burlington, MA). TF-1, MOLM13 and MDS-L were cultured in RPMI 1640 (Thermo Fischer Scientific, Waltham, MA) with 10% FBS and F36P, SET2 and SKM-1 with 20% FBS with the addition of GM-CSF for TF-1 and F36P (2 ng/ml and 20 ng/ml respectively; PeproTech). All the cell lines were cultured with 2mM L-glutamine, penicillin (100 U/ml) and streptomycin (100μg/ml) at 37 in 5% CO2. Doxycycline was purchased from Sigma Aldrich (D9891) and was diluted in PBS (Thermo Fischer Scientific, Waltham, MA). Gemtuzumab (mylotarg) was purchased from MedChem Express (#HY-109539).
ADCs generation
The anti-CD33/CD123/humanCCRL2/mouseCCRL2 and isotope controls (IgG2a and IgG2b) ADCs were developed by conjugating the anti-CD33 (Santa Cruz #374450), anti-CD123 (BD Biosciences #554527), anti-humanCCRL2 antibody (BioLegend #358302), anti-mouseCCRL2 antibody (BioLegend #114002), IgG2a and IgG2b isotype control antibodies (BioLegend # 400502/ 400602) with PBD using a dipeptide linker (SG3249 (MedChemExpress Cat. No.: HY-128952). was done by reducing monoclonal antibodies using 5x molar excess Tris(2-carboxyethyl) phosphine hydrochloride (TCEP, ThermoFisher Scientific, Cat. No: 20490) followed by TCEP removal using Zeba Spin columns (ThermoFisher Scientific, Cat No: 89882). Subsequently, antibodies were reacted with 5x molar excess SG3249 in 10% DMSO at room temperature. Excess unreacted drug linkers were subsequently removed by buffer exchange into PBS using Zeba Spin columns. ADC drug conjugation was analyzed by hydrophobic interaction chromatography (HIC) using Agilent 1260 Infinity I LC system as previously described. Baseline correction and analysis of HPLC spectra was performed using the OriginPro v10.1.5.132. The HPLC methods and drug-antibody ratio (DAR) calculations were described before(20).
MTT assay
TF-1, F36P, K562, THP1, SET2, MOLM13, MV4–11, MDS-L and SKM1 cells were plated in 96-well plates (7,500 cells per well) and treated for 6 days with anti-CCRL2, anti-CD33, anti-CD123, gemtuzumab and IgG2a antibodies. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay (11465007001, Roche Diagnostics, Mannheim, Germany) was conducted according to the manufacturer’s instructions. Absorbance was measured at 570 nm. Calculation of cell viability by 100x (absorbance of sample/average absorbance of untreated control) for the respective dose for each cell line.
Flow cytometry analysis
Healthy control (HC) samples were stained with FITC-conjugated anti-human CD34 (BioLegend, #343603), BV605-conjugated anti-human CD38 (BioLegend, #303531), BV421-conjugated anti-human CD71(Biolegend, #334121), and APC-conjugated anti-human CD235a (#349113). Cell lines were stained with PE-conjugated anti-CCRL2 (BioLegend, #358303), APC-conjugated anti-CD123 (BioLegend #973704), and PE-conjugated anti-CD33 (BioLegend #303403). Primary samples were stained with BV510-conjugated anti-human CD45 (BioLegend, #103137) and PE-conjugated anti-CCRL2. Cell line and patient-derived xenograft samples were stained with PE-Cy7-conjugated anti-mouse CD45 (BioLegend #103113), and BV510-conjugated anti-human CD45 (BioLegend, #103137). Gating was based on an unstained control. For the assessment of apoptosis, cells were stained with 7AAD (BioLegend, #420403). Following staining, analysis was performed using BD LSR II (BD Biosciences). Mean fluorescence intensity (MFI) was measured for each marker using FlowJo analysis software version 10.0.8(FlowJo, Ashland, CO, USA).
CCRL2 knockout (KO)
Lentiviral vectors expressing CCRL2-targeting sgRNA (pLV [CRISPR]-hCas9:T2A: Puro-U 6 > hCCRL2[gRNA#162], pLV [CRISPR]-hCas9:T2A: Puro-U6 > hCCRL2 [gRNA#177]) or empty pLKO.1-puro lentiviral vector (pLV [CRISPR]-hCas9/Puro-U6 > Scramble_gRNA1) was transfected into 293T cells using Lipofectamine 2000 (Thermo Fischer Scientific) for lentiviral supernatant production.TF-1, F36P and SET2 cells were incubated with the viral supernatant and polybrene (8μg/ml; MilliporeSigma) for transduction. 48 hours later, cells were treated with puromycin (2μg/ml for TF-1, 1.5 μg/ml for F36P and 0.75 μg/ml for SET2) for 3–4 days for resistant cells selection.
Colony formation assay
Clonogenic assays were conducted as previously detailed(15, 16). Cells were counted and resuspended at a density of 3000 cells/ml in methylcellulose-based media. Following around two weeks of incubation at 37°C in 5% CO2, counting of colony forming units was performed under bright-field microscopy. A colony was defined as a cell aggregate of > 50 cells.
Patients and sample processing
All specimens were obtained by the Johns Hopkins Kimmel Cancer Center Specimen Accessioning Core. In accordance with the Declaration of Helsinki and under a research protocol approved by the Johns Hopkins Institutional Review Board, informed consent was procured from all donors before specimen collection. Isolation of CD34 + cell subsets was performed using the CD34 MicroBead kit (Miltenyi Biotec) as before. Samples were collected from marrow aspirations of multi-hit TP53-mutated and wild-type MDS/AML or de novo AML patients. Control marrow was collected as excess material after harvesting healthy donors for allogeneic bone marrow transplantation. ADCs (20 or 40 ng/ml for TP53-mutated or TP53 wild-type MDS/AML primary cells and 100 ng/ml for HC MNCs or CD34 + cells) were added at day 0 to MNCs or sorted CD34 + cells. At day 5, apoptosis was assessed by 7AAD staining and, differentiation was assessed by CD34, CD38, CD71, CD235a staining for HC samples. Clonogenicity was assessed by plating 2,000 cells in methylcellulose-based medium (StemCell #04434).
Treatment of C57BL/6 mice
Total 14 8–10 weeks old C57BL/6 mice were treated intravenously with 1 mg/kg anti-mouse CCRL2 ADC or conjugated IgG2b. Complete blood counts and chemistry panels were analyzed before treatment and 4 weeks later. Weight was measured every week. Mice were harvested at 4 weeks post-treatment and kidney and spleen weights were measured.
Cell line xenografts
Luciferase + TF-1, SKM1 and F36P cells were injected to 8–10-week-old NOD.Cg-Prkdc^scid^ Il2rg^tm1Wjl^/SzJ (NSG) female mice (10^6^ cells per mouse) (14, 15). Using IVIS spectrum in vivo imaging system, bioluminescence signal was measured at different timepoints. At day 10 or 15, mice were randomized to receive either conjugated IgG2a or anti-CCRL2 ADC (0.75 mg/kg for TF-1, 1 mg/kg for SKM1 and F36P, intravenously). Among F36P engrafted mice, 5 vs 5 mice were sacrificed on day 35 (after one dose of treatment) to assess disease burden and 4 vs 4 mice were monitored until day 65. Survival of the mice was assessed by day 65 for TF-1, 50 for SKM1 and 75 for F36P. At that time, all remaining mice were euthanized, and the percentage of human CD45 + cells was assessed in bone marrow, spleens or livers by flow cytometry. IACUC guidelines were followed.
Patient-derived multi-hit TP53-mutated MDS/AML xenografts
MNCs collected from marrow aspirates of multi-hit TP53-mutated MDS/AML were injected to sub-lethally irradiated (1.5 Gy) 8–10-week-old NOD.Cg-Prkdc^scid^ Il2rg^tm1Wjl^ Tg (CMV-IL3, CSF2, KITLG)1Eav/MloySzJ (NSGS) female mice (5×10^5^ cells/mouse). Bone marrow aspirates and peripheral blood were collected from mice at day 30 and based on disease burden, engrafted mice (≥ 2% human CD45 + cells in bone marrow) were randomized to receive conjugated IgG2a or anti-CCRL2 ADC (1 mg/kg). At day 55, peripheral blood, spleens, livers and bone marrows were collected from mice to assess disease burden by flow cytometry and immunohistochemical analysis. IACUC guidelines were followed.
Immunohistochemistry
Following preparation of paraffin bone marrow, spleen and liver sections, slides were prepared and stained using hematoxylin and eosin (H&E).
Statistical Analysis
Statistical analysis was performed by using GraphPad Prism (GraphPad Software, La Jolla, CA). Mann-Whitney test was performed to assess statistical significance when comparing two groups. One-way analysis of variance (ANOVA) was performed for the comparisons of three or more groups. Kaplan-Meier analysis and mantle Cox log rank test was performed for comparisons of mice survival. Standard deviation was used to assess centrality and dispersion.
Results
Anti-CCRL2 ADC is cytotoxic against MDS/AML cell lines
To identify the best payload for the development of an ADC with high activity against high-risk MDS/AML, five commonly used ADC payloads (DM1, MMAE, Exatecan, SN38, PBD) were compared and pyrrolobenzodiazepine (PBD) showed the highest cytotoxicity against the erythroleukemic/ TP53-mutated TF-1 cells, which was not affected by CCRL2 expression based on the comparison of CCRL2 wild-type (WT) or knock-out (KO) cells (Supplementary Figs. 1A-B). The (PBD) dimer (SG3199)(21)-conjugated ADC loncastuximab has received FDA approval for high-grade B-cell lymphomas treatment(22, 23). Thus, an anti-CCRL2 ADC was developed by conjugating a monoclonal anti-human CCRL2 antibody, with PBD. A PBD-conjugated IgG2a antibody (IgG2a ADC) was used as control. Conjugations were confirmed by high-performance liquid chromatography (Supplementary Fig. 1C, D) showing a drug-antibody ratio (DAR) of ~ 3.7 (Supplementary Table 1).
To assess anti-CCRL2 ADC selective efficacy against cells expressing CCRL2, CCRL2 WT and KO TF-1, F36P and SET2 (16) cells, were treated with increasing doses (0–300 ng/ml) of ADCs for 6 days. The anti-CCRL2 ADC had a significantly higher cytotoxicity against WT compared to KO cells and compared to conjugated IgG2a (Fig. 1A–C). To further assess CCRL2-selective activity doxy-inducible CCRL2 TF-1 cells (iTF-1) which are genetically engineered TF-1 cells to express CCRL2 under doxycycline (15), were used. The activity of anti-CCRL2 ADC was significantly higher when cells were treated with doxycycline compared to untreated ones (Fig. 1D).
Treatment with 20 or 40 ng/ml anti-CCRL2 ADC induced apoptosis in TF-1, F36P, SKM1 and MDS-L cells, compared to treatment with 40 ng/ml conjugated IgG2a (Fig. 1E). Similarly, 10 and 20 ng/ml anti-CCRL2 ADC suppressed clonogenicity in these cells compared to treatment with the same doses of conjugated IgG2a (Fig. 1F).
Collectively, anti-CCRL2 ADC shows CCRL2-selective cytotoxicity against MDS/AML cell lines in vitro(22, 24, 25).
Anti-CCRL2 ADC shows higher cytotoxicity against MDS/AML cell lines compared to gemtuzumab and PBC-conjugated ADCs targeting CD33 and CD123
Next, to compare the efficacy of ADC targeting CCRL2 with the efficacy of ADCs against other targets, PBD-conjugated ADCs targeting the well described AML targets CD33, and CD123 were developed (Supplementary Figs. 1E-F). The cytotoxicity of the calichemicin-conjugated ADC gemtuzumab and the PBD-conjugated anti-CD33, -CD123, -CCRL2 ADCs were assessed in AML cell lines derived from patients with de novo/TP53-wild type AML (MOLM13, OCI-AML3 and MV4–11) (Fig. 2A–C) and MDS/AML cell lines (TF-1 - erythroleukemia, F36P – MDS-related erythroleukemia, and SKM1 – TP53-mutated MDS/AML) (Fig. 2D–F). Consistently, with our previously published data, CCRL2 expression is significantly higher in MDS/AML cell lines with erythroleukemic features and TP53 mutations compared to AML cell lines derived from patients with de novo/TP53-wild type AML (Supplementary Fig. 1G).
AML cell lines derived from de novo/TP53-wild type AML were overall more sensitive to ADCs treatment compared to MDS/AML cell lines (Fig. 2A–C). Particularly, the activity of gemtuzumab, and PBD-conjugated ADCs targeting CD33 and CD123 was found to be significantly decreased in MDS/AML cell lines compared to AML cell lines derived from de novo/ TP53-wild type AML. The anti-CCRL2 ADC showed overall similar or lower cytotoxicity compared to Gemtuzumab and PBD-conjugated ADCs targeting CD33 and CD123 in the AML cell lines derived from de novo/ TP53-wild type AML (Fig. 2A, C). However, within the MDS/AML cell lines, the anti-CCRL2 ADC showed the highest cytotoxicity in TF-1 and F36P cells and the second to highest cytotoxicity in SKM1 cells (Fig. 2D–F).
Overall, the activity of anti-CCRL2 ADC appears to be higher compared to other ADCs against MDS/AML cell lines.
Anti-CCRL2 ADC induces apoptosis and suppresses clonogenicity in MDS/AML primary cells
Next, the activity of the anti-CCRL2 ADC in primary mononuclear cells from MDS/AML patients was analyzed. Based on our published data supporting higher expression of CCRL2 in MDS/AML, TP53-mutated disease and myeloid neoplasms with erythroid features compared to de novo AML (14, 16), 4 patients with de novo/TP53-wild-type AML, 4 patients with TP53-wild-type MDS-related AML and 10 patients with multi-hit TP53-mutated (complex karyotype with TP53 mutation with variant-allele frequency ≥ 50%) MDS/AML including 3 individuals with acute erythroid leukemia (Supplementary Tables 2, 3 and Fig. 3A) were included. Cells were treated with 20 or 40 ng/ml anti-CCRL2 ADC or 20 ng/ml conjugated IgG2a for 5 days. Treatment with 10 and 20 ng/ml anti-CCRL2 increased apoptosis and suppressed cells clonogenicity compared to conjugated IgG2a (Fig. 3B). Fluorescent in situ hybridization (FISH) of isolated colonies confirmed the presence of 17p and 5q deletions and trisomy 8 in isolated cells (Supplementary Fig. 2). To assess if CCRL2 expression is associated with the cytotoxicity of the anti-CCRL2 ADC, the expression of CCRL2 in the surface of primary blasts was measured by flow cytometry (Fig. 3C). Consistently with our previously reported results(14, 16), acute erythroid leukemia blasts express the highest levels of CCRL2 and TP53-mutated MDS/AML blasts express relatively higher levels of CCRL2 compared to de novo AML (Fig. 3C). Linear regression showed a positive correlation between CCRL2 expression and anti-CCRL2 ADC apoptotic effect defined as the ratio of apoptosis induced by 40 ng/ml anti-CCRL2/apoptosis induced by 40 ng/ml IgG2a (Fig. 3D).
Taken together, our results support that anti-CCRL2 ADC promotes apoptosis and suppresses the clonogenicity of primary MDS/AML cells. While de novo AML samples demonstrated similar cytotoxicity compared to MDS/AML and TP53-mutated samples, higher CCRL2 expression, is associated with better response. These findings suggest that the observed anti-leukemic activity in TP53 mutated MDS/AML and particularly erythroid leukemias is likely associated with higher expression of the target and not cell-intrinsic sensitivity to the ADC.
Anti-CCRL2 ADC does not affect healthy hematopoietic cells and does not cause systemic toxicity in vivo
To assess the effect of anti-CCRL2 ADC on healthy hematopoietic cells, CD34 + cells sorted from bone marrows of 3 independent healthy donors (Supplementary Table 2) were treated with 100 ng/ml conjugated IgG2a or anti-CCRL2 ADC, which is a substantially higher dose compared to the doses selected to treat primary MDS/AML samples for 5 days. Treatment with anti-CCRL2 ADC did not induce apoptosis (Fig. 4A), affect differentiation (Fig. 4B) or suppress clonogenicity (Fig. 4C) of healthy CD34 + cells. Given that CCRL2 is overexpressed in leukemias with erythroid differentiation and its deletion decreases the survival and clonogenicity of erythroleukemic cells(16), the effect of anti-CCRL2 ADC on healthy erythroid progenitors was assessed. Mononuclear cells from 3 additional healthy bone marrow donors were treated with 100 ng/ml anti-CCRL2 or conjugated IgG2a for 5 days. Treatment with anti-CCRL2 ADC did not alter the percentages of erythroid progenitors CD71 + CD235a-, CD71 + CD235a + or CD71-CD235a) (Fig. 4D).
To further characterize the safety profile of this agent in vivo, an anti-mouse CCRL2 ADC was developed (Supplementary Fig. 3A). Healthy 6–8 weeks C57BL/6 female mice were treated intravenously with 1 mg/kg anti-CCRL2 ADC or conjugated IgG2b (7 mice per group) (Fig. 4E). No significant weight changes or systemic toxicities were observed following treatment (Fig. 4F) and the weight of spleens and kidneys were not affected by treatment at 4 weeks following injection (Supplementary Fig. 3B). Consistently, no significant alterations of the mice complete blood counts (Fig. 4G, Supplementary Fig. 3C) or chemistry panels (Fig. 4H, Supplementary Fig. 3D) were found.
Collectively, our results support that anti-CCRL2 ADC has a relatively good safety profile without prominent effect on healthy hematopoiesis.
Anti-CCRL2 ADC suppresses leukemic growth and improves the survival in TP53-mutated MDS/AML cell line xenografts
To test the in vivo efficacy of one dose of anti-CCRL2 ADC in TP53-mutated MDS/AML models, TF-1 and SKM1 xenografts were treated. Luciferase + TF-1 cells were intravenously injected in NSG mice. On day 15, to ensure balanced baseline tumor burdens, mice were stratified into groups with comparable bioluminescence signals and received anti-CCRL2 ADC or conjugated IgG2a (0.75 mg/kg, one dose) (Fig. 5A). Mice treated with anti-CCRL2 ADC showed suppressed leukemic growth (Fig. 5B) and improved survival compared to those treated with conjugated IgG2a (median survival 47 vs 37 days) (Fig. 5C). Luciferase + SKM1 cells were intravenously injected in NSG mice, which were randomized based on their bioluminescence signal on day 11 to anti-CCRL2 ADC or conjugated IgG2a (1 mg/kg, one dose) (Fig. 5D). Treatment with anti-CCRL2 ADC suppressed leukemic growth (Fig. 5E) and significantly prolonged the mice survival compared to those treated with conjugated IgG2a (median survival 39 vs 9.5 days) (Fig. 5F). Of note, mice engrafted with SKM1 cells exhibit particularly enlarged livers at the time of their death. The mice treated with anti-CCRL2 ADC had significantly lower liver weights compared to those treated with IgG2a (Fig. 5G).
Next, the effect of multiple anti-CCRL2 ADC doses was tested in the least aggressive TP53-mutated MDS/AML F36P xenograft model. At day 15 following intravenous injection of Luciferase + F36P cells, mice received 1 mg/kg anti-CCRL2 ADC or conjugated IgG2a (Fig. 6A, Supplementary Fig. 4A). At day 35, mice were sacrificed, and those treated with anti-CCRL2 ADC had significantly lower percentage of human CD45 + cells (hCD45+%) in their bone marrows compared to those (Fig. 6B). Another group of F36P xenografts received two doses of 1 mg/kg of anti-CCRL2 ADC or conjugated IgG2a at days 15 and 35 (Fig. 6C). Mice treated with anti-CCRL2 ADC demonstrated suppressed leukemic growth (Fig. 6D), improved survival (median survival 55 vs 38.5 days) (Fig. 6E) and decreased hCD45+% in their bone marrows compared (Fig. 6F) to those treated with conjugated IgG2a.
Taken together, these results support that treatment with anti-CCRL2 ADC suppresses the leukemic growth of TP53-mutated MDS/AML cell line xenografts.
Anti-CCRL2 ADC suppresses the leukemic growth in multi-hit TP53-mutated MDS/AML patient-derived xenografts
Next, the efficacy of anti-CCRL2 ADC in patient-derived xenografts was assessed. Mononuclear cells from 5 TP53-mutated MDS/AML patients (Supplementary Table 4) were injected in sub-lethally irradiated (1.5 Gy) NSGS mice. Engraftment was assessed on day 30 by peripheral blood and bone marrow aspirate flow cytometry analysis (Fig. 7A). Three out of the 5 patient samples were successfully engrafted (≥ 2% hCD45 + in bone marrow) in total 11 mice (Fig. 7A). To ensure balanced baseline tumor burdens, mice were stratified into groups with comparable hCD45+% in their bone marrows, received anti-CCRL2 ADC or conjugated IgG2a (intravenously, 1 mg/kg) and harvested at day 55 (Fig. 7A). The mice who were treated with anti-CCRL2 ADC showed a reduction or stabilization of hCD45+% in their peripheral blood (Fig. 7B) or their bone marrow (Fig. 7C) compared to those treated with conjugated IgG2a. The hCD45+% in the bone marrow was not significantly different between the two groups (Supplementary Fig. 4B) but given the significant variation of hCD45+% at baseline, the hCD45+% fold change was assessed and was found significantly lower in mice treated with anti-CCRL2 ADC compared to those treated with conjugated IgG2a (Fig. 7D). Similarly, mice treated with anti-CCRL2 ADC had significantly lower hCD45+% in their spleens compared to those treated with conjugated IgG2a (Fig. 7E).
Pathologic analysis of mouse tissues revealed that mice treated with conjugated IgG2a had more advanced disease involvement in their marrows, spleens and livers compared to those treated with anti-CCRL2 ADC (Fig. 7F). An MDS/AML involvement score was developed (Supplementary Fig. 4C) and mice treated with anti-CCRL2 ADC had significantly lower scores compared to those treated with conjugated IgG2a (Fig. 7G).
Collectively, these results suggest that treatment with anti-CCRL2 ADC suppresses leukemic growth in multi-hit TP53-mutated patient-derived MDS/AML xenografts.
Discussion
Patients with high-risk MDS/AML including those with TP53 mutations and deletions continue to exhibit relatively poor outcomes due to the lack of effective therapies and high incidence of treatment resistance(1–3). ADCs including the PBD-conjugated loncastuximab, have prominent activity in hematologic malignancies (8, 9, 22, 23), but the currently available ADCs such as the anti-CD33 ADC gemtuzumab, show limited activity in high-risk MDS/AML and TP53-mutated disease (10). We developed an anti-CCRL2 ADC using the PBD payload and showed a significant single-agent anti-leukemic effect in various MDS/AML models.
Our studies demonstrated that cell lines derived from patients with MDS/AML or AML with erythroid features are generally less sensitive to gemtuzumab and PBD-conjugated ADCs targeting CD33 and CD123, compared to AML cell lines from patients with de novo or good- to intermediate-risk disease. This finding aligns with the clinical responses observed in high-risk MDS/AML patients treated with gemtuzumab(10). In contrast, the anti-CCRL2 ADC exhibited consistent efficacy across the different cell line subtypes, demonstrating higher activity against cell lines derived from patients with TP53 mutated MDS/AML or AML with erythroid features compared to gemtuzumab and PBD-conjugated ADCs targeting CD33 and CD123. These results suggest that the marked upregulation of CCRL2 in high-risk MDS/AML and TP53-mutated cells may offset their inherent resistance to drug toxin-mediated cell killing.
Analysis of anti-CCRL2 ADC activity in primary samples confirmed that this agent induces apoptosis and suppresses the clonogenic potential of MDS/AML cells. MDS/AML and TP53-mutated samples showed overall similar sensitivity to anti-CCRL2 ADC compared to de novo AML samples with erythroleukemia samples showing the highest sensitivity. This result is consistent with our findings in MDS/AML cell lines demonstrating consistent activity of the anti-CCRL2 ADC to de novo and MDS/AML cell lines. Notably, there was a positive association between CCRL2 expression and the apoptotic activity induced by the agent, consistent with findings reported for other ADCs targeting CD33(26–28). Our group recently demonstrated that TP53-mutated MDS/AML and AML with erythroid features exhibit the highest levels of CCRL2 expression across the spectrum of MDS/AML subtypes(16). These findings support the significant anti-leukemic effect of anti-CCRL2 ADC in TP53-mutated MDS/AML and particularly erythroleukemic samples is likely associated with the prominent overexpression of the target in these subsets and not with their cell intrinsic sensitivity to the released toxin. On the contrary, de novo AML samples are probably more sensitive to toxin-mediating cell killing based on our cell lines data, counterbalancing their relatively low CCRL2 expression.
PBD-conjugated antibody-drug conjugates (ADCs) targeting CD33 and CD123 showed promising preclinical efficacy in high-risk AML(29, 30). However, early-phase clinical trials revealed cytopenias and hepatotoxicity, which hindered further clinical development. Notably, not only gemtuzumab but also bispecific antibodies and CAR-T cells targeting CD33, have been associated with cytopenias(31–33). This suggests that these adverse effects may be related to the expression of CD33 and CD123 on the surface of healthy hematopoietic cells. While transient elevations in liver enzymes have been observed with PBD payloads, there have been no reports of fatal hepatotoxicity(22, 23). On the contrary, healthy stem and progenitor cells express minimal CCRL2 levels(14, 15). Consistently, anti-CCRL2 ADCs demonstrated minimal impact on healthy hematopoietic cells, and did not induce systemic toxicity or significant changes in blood counts or chemistry panels in C57BL/6 mice, similarly to recent findings from our group(34). Should cytopenias or hepatotoxicity arise with anti-CCRL2 ADCs, fractionated dosing may be considered, as this approach has enabled the safe and effective use of gemtuzumab.
Our in vivo studies demonstrated that the anti-CCRL2 ADC effectively suppressed leukemic growth in cell line xenograft models, resulting in improved survival of treated mice. Notably, two doses of the agent administered 20 days apart were well tolerated by NSG mice and led to a marked reduction in disease burden. Furthermore, bone marrow aspirates collected from NSGS mice engrafted with TP53-mutated MDS/AML cells enabled randomization to treatment with either anti-CCRL2 ADC or an isotype control. This approach revealed significant anti-leukemic activity of the anti-CCRL2 ADC. However, elimination of TP53-mutated MDS/AML cells was not achieved, suggesting that combination therapeutic strategies may be necessary to obtain clinically meaningful responses.
Further studies in syngeneic MDS/AML models are required to solidify the efficacy and safety of anti-CCRL2 ADC, but this agent appears to be a promising candidate for combination therapies for high-risk MDS/AML including TP53-mutated disease especially given the lack of effective antibody-based therapies in these disease subsets. Similarly, conjugating antibodies targeting other surface antigens that are particularly overexpressed in high-risk MDS/AML and show very low expression in healthy hematopoietic cells, with PBD may be an attractive strategy to improve the outcomes of patients with these neoplasms.
Supplementary Files
This is a list of supplementary files associated with this preprint. Click to download.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Kröger N. Treatment of high-risk myelodysplastic syndromes. Haematologica. 2025;110(2):339–49.39633555 10.3324/haematol.2023.284946 PMC 11788630 · doi ↗ · pubmed ↗
- 2Daver NG, Maiti A, Kadia TM, Vyas P, Majeti R, Wei AH, TP 53-Mutated Myelodysplastic Syndrome and Acute Myeloid Leukemia: Biology, Current Therapy, and Future Directions. Cancer Discov. 2022;12(11):2516–29.36218325 10.1158/2159-8290.CD-22-0332 PMC 9627130 · doi ↗ · pubmed ↗
- 3Hochman MJ, Othus M, Hasserjian RP, Ambinder A, Brunner A, Percival MM, Prognostic impact of secondary versus de novo ontogeny in acute myeloid leukemia is accounted for by the European Leukemia Net 2022 risk classification. Leukemia. 2023;37(9):1915–8.37524919 10.1038/s 41375-023-01985-y PMC 10457181 · doi ↗ · pubmed ↗
- 4Fu Z, Li S, Han S, Shi C, Zhang Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct Target Ther. 2022;7(1):93.35318309 10.1038/s 41392-022-00947-7PMC 8941077 · doi ↗ · pubmed ↗
- 5Paul S, Konig MF, Pardoll DM, Bettegowda C, Papadopoulos N, Wright KM, Cancer therapy with antibodies. Nat Rev Cancer. 2024;24(6):399–426.38740967 10.1038/s 41568-024-00690-x PMC 11180426 · doi ↗ · pubmed ↗
- 6Calabretta E, Hamadani M, Zinzani PL, Caimi P, Carlo-Stella C. The antibody-drug conjugate loncastuximab tesirine for the treatment of diffuse large B-cell lymphoma. Blood. 2022;140(4):303–8.35580172 10.1182/blood.2021014663 PMC 9335500 · doi ↗ · pubmed ↗
- 7Kantarjian HM, De Angelo DJ, Stelljes M, Martinelli G, Liedtke M, Stock W, Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N Engl J Med. 2016;375(8):740–53.27292104 10.1056/NEJ Moa 1509277 PMC 5594743 · doi ↗ · pubmed ↗
- 8Freeman SD, Thomas A, Thomas I, Hills RK, Vyas P, Gilkes A, Fractionated vs single-dose gemtuzumab ozogamicin with determinants of benefit in older patients with AML: the UK NCRI AML 18 trial. Blood. 2023;142(20):1697–707.37595359 10.1182/blood.2023020630 PMC 10667325 · doi ↗ · pubmed ↗