Impact of Fertilisation on the Bacterial Core Microbiome of Grassland Soils: Abundance in the Field and Growth In Vitro
Rostand R. Chamedjeu, Kunal Jani, Karoline Jetter, Kerstin Wilhelm, Patrick Schäfer, Lena Wilfert, Simone Sommer, Christian U. Riedel

TL;DR
This study shows how different fertilization methods, especially pig slurry, affect the bacterial makeup of grassland soil and how these effects can be studied in lab experiments.
Contribution
The study demonstrates that culturable soil bacteria can be used to replicate field observations in controlled lab settings under different fertilization regimes.
Findings
Pig slurry fertilization caused the most significant changes in the bacterial core microbiome composition.
Bradyrhizobium japonicum growth was inhibited by pig slurry, while Nocardioides albus and Solirubrobacter pauli grew better in its presence.
Culturable bacteria mirrored field abundance patterns in in vitro experiments, validating lab-based microbiome studies.
Abstract
Anthropogenic activities may have profound impacts on the soil microbiome with consequences for soil health, agriculture and food production. Here, we investigated the impact of different fertilisation regimes on the composition of the bacterial soil microbiome in grassland ecosystems by 16S rRNA gene amplicon sequencing and in vitro growth experiments with culturable representatives of the bacterial core microbiota. We observed a large proportion of taxa shared across fertilisation regimes without significant differences in their evenness, but shifts in the composition of the bacterial core microbiome by fertilisation. These effects were most pronounced for fertilisation with pig slurry (PS). Analysis of microbiome multivariable association with linear models identified bacterial biomarker taxa for different fertilisation regimes. This enabled the selection of several culturable…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 FIGURE 1
FIGURE 1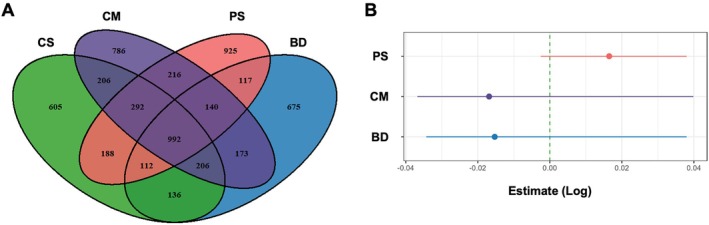 FIGURE 2
FIGURE 2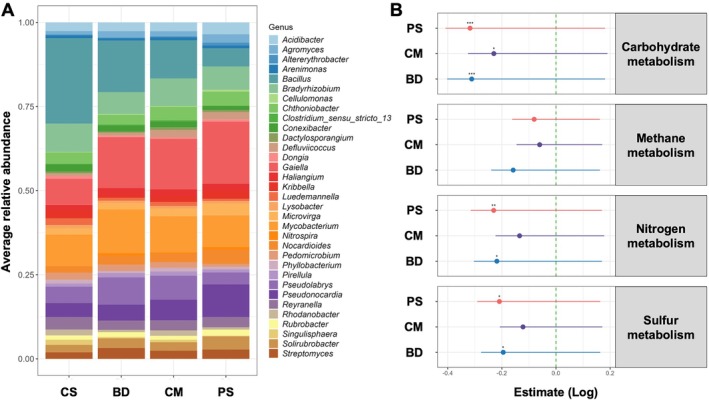 FIGURE 3
FIGURE 3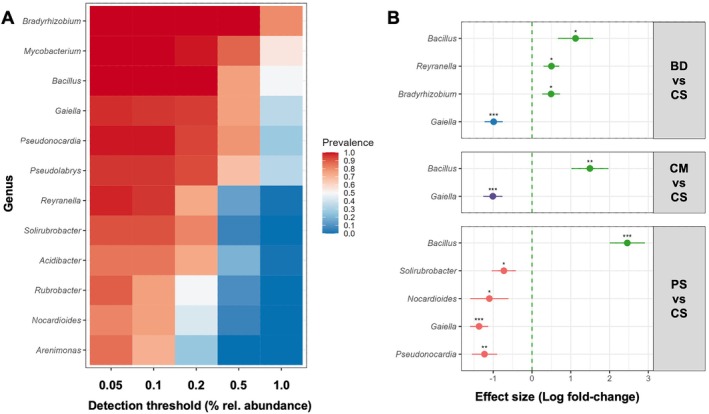 FIGURE 4
FIGURE 4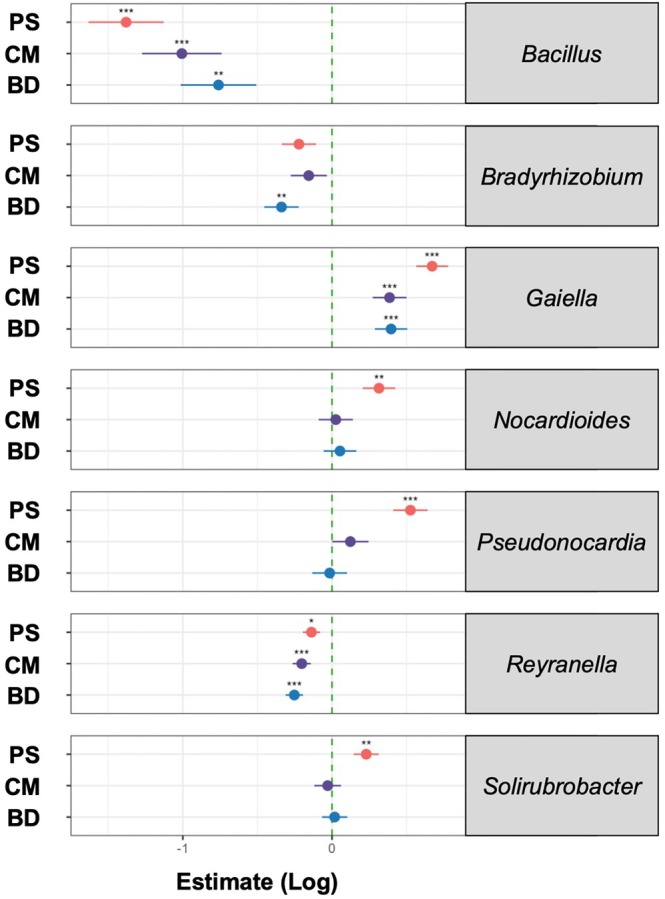 FIGURE 5
FIGURE 5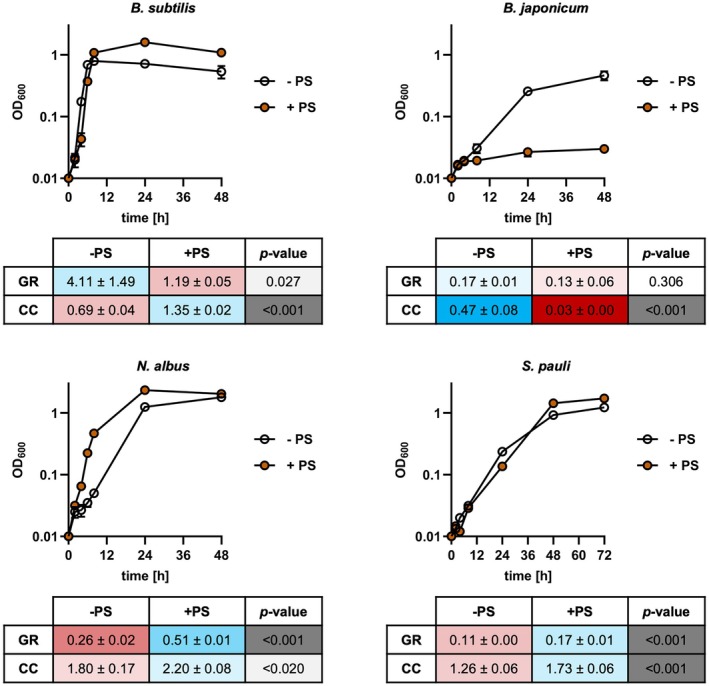 FIGURE 6
FIGURE 6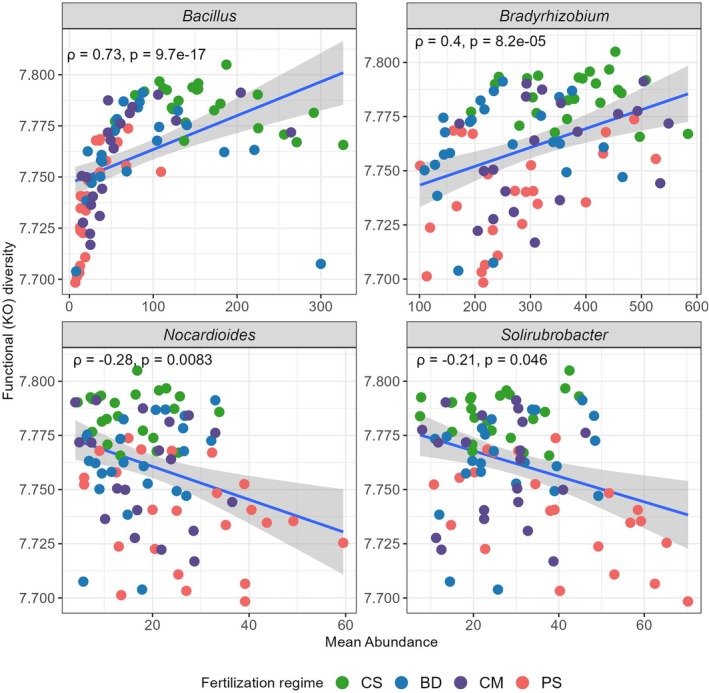 FIGURE 7
FIGURE 7- —Baden‐Württemberg Stiftung10.13039/100008316
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSoil Carbon and Nitrogen Dynamics · Microbial Community Ecology and Physiology · Gut microbiota and health
Introduction
1
Soil provides numerous ecosystem functions and services, including plant growth, nutrient cycling, carbon sequestration, and water purification (Timmis and Ramos 2021; Kopittke et al. 2024). As such, soil health is crucial for sustainable agriculture, reducing the risk of soil degradation and supporting resilience against environmental changes (Kopittke et al. 2024; Raupp et al. 2024). The microbiome is a major biological driver of ecosystem functions and services of soil, and its activity is influenced by abiotic factors, such as soil physico‐chemical properties, as well as external factors like agricultural land use and fertilisation (Bissett et al. 2011; Philippot et al. 2024). In addition, soil microorganisms play a central role in terrestrial biogeochemistry, driving the cycling of soil organic matter, which represents the largest primary source of plant nutrients (Fierer 2017; Yadav et al. 2021). Previous studies have demonstrated a positive correlation between the diversity of the soil microbiome and a range of ecosystem functions including nutrient cycling, decomposition of soil organic matter, plant development, growth and protection against pathogens (Delgado‐Baquerizo et al. 2020; Trivedi et al. 2022). In addition, the soil microbiome also represents an important genetic resource for global health (Lehmann et al. 2020; Wood and Blankinship 2022). For example, many soil microbes harbour biosynthetic gene clusters related to the production of antimicrobials, and their diversity is of relevance to address the current crisis of antimicrobial resistance (de Castro et al. 2014; Ma et al. 2023; Zhang et al. 2024).
Gradual and persistent anthropogenic impacts can cause shifts in the diversity and activity patterns of the soil microbiome and thus may have consequences for the functional stability of the soil ecosystem (Siebert et al. 2019; Thakur et al. 2020). Land‐use intensification represents one of the major anthropogenic factors that alter soil biodiversity and affect ecosystem processes (Matson et al. 1997; Babin et al. 2021). In agricultural ecosystems under intensified human land use, loss in biodiversity and decline in ecosystem functioning are associated with significant soil degradation, loss of productivity and increased greenhouse gas emissions (Hurni et al. 2015). For example, conventional tillage is deleterious for soil physicochemical and biological properties (Congreves et al. 2017), which may have consequences in soil microbiome structures. It is also known that different management practices in agroecosystems are associated with different diversity patterns of the soil microbiome. However, the effects of different organic fertilisation regimes on microbial diversity in soil and the consequences on ecosystem functions are currently poorly understood impeding our ability to develop adapted management systems for sustainable agricultural land use.
A deeper understanding of soil microbial communities, often referred to as the microbiome or microbiota, is essential to comprehend their contribution to these vital ecosystem functions. While the terms microbiome and microbiota have partially overlapping definitions and are often used interchangeably, they have distinct definitions (Berg et al. 2020). The microbiome is typically referred to as “a characteristic microbial community occupying a reasonably well‐defined habitat with distinct physio‐chemical properties,” encompassing not only the microorganisms but also their metabolic activities. By contrast, microbiota refers specifically to the assembly of microorganisms across different kingdoms, including Prokaryotes (Bacteria, Archaea) and Eukaryotes (e.g., Protozoa, Fungi, and Algae). This distinction is important for understanding the complex dynamics within soil ecosystems and how they may be influenced, for example by agricultural practices such as fertilisation.
One approach to evaluate the impact of various factors on the condition and functions of an ecosystem is to analyse biomarker organisms or communities and their responses (Gerhardt 2002). More recently, ‘next‐generation biomonitoring’ has been used to analyse changes in diverse ecosystems including soil by sequencing entire genetic information using next‐generation technologies and machine learning algorithms (Bohan et al. 2017; Fierer 2017; Cordier et al. 2019, 2021). In this study, we investigate the impact of different organic fertilisation regimes on the composition of the bacterial soil microbiome in grassland ecosystems. We hypothesized that different organic fertilisation regimes have distinct effects on the composition and functional diversity of the bacterial grassland soil microbiome by enriching and/or depleting specific taxa. Focusing on the bacterial components of the soil microbiome, we sequenced 16S rRNA gene amplicons to identify representative species that may then be used as biomarkers for fertilisation status. Additionally, we sought to test whether the effects of fertilisation can be replicated in in vitro growth experiments with representative, culturable type species of these biomarkers. This approach may help to better understand the impact of organic fertilisation on soil microbial dynamics and improve predictions of the responses of agricultural ecosystems to anthropogenic disturbances. Furthermore, this may support the development of more robust biomonitoring methods and promote sustainable management of ecosystem services in grassland ecosystems affected by intensive land use or other environmental stresses.
Materials and Methods
2
Study Sites and Sampling Design
2.1
Soil samples were collected from grassland ecosystems on the Swabian Alb, Germany (Figure 1: Graphical abstract). The soils in this region are primarily loamy and silty clays with high skeletal content. The farming systems in the study area follow specific regimes of mowing, grazing, and organic fertilisation. The experimental design included study sites managed for several years by four different fertilisation regimes (n = 5–6 study sites per management regime). Three groups of study sites were intensively managed with several rounds of fertilisation and mowing per year. The study sites were either fertilised with pig slurry (PS), cow manure (CM), or biogas digestate (BD). Control soil (CS) was obtained from a fourth group of sites mowed only once a year and fertilised exclusively by occasional grazing by flocks of sheep (1–2× per year). Except for PS sites, all sites are part of the Biodiversity Exploratories “Schwäbische Alb” (https://www.biodiversity‐exploratories.de/en/). Soil samples were collected between May and July 2022 from the top‐soil at 0–20 cm depth. Per site, four samples were collected from randomly selected spots. Before further processing, stones and plant debris were removed by passing samples through a sieve (mesh size 0.1 cm). Samples were immediately placed in a cooling box, transported to the laboratory, and stored at −80°C until further processing. In total, 92 samples (4 regimes, 5–6 sites per regime, 4 biological replicates per site) were obtained for 16S rDNA gene amplicon sequencing.
Workflow of the presented study with a map of the study sites for field sample collection, experimental setup, sample processing, 16S rRNA gene amplicon sequencing, analysis of the bacterial microbiome, and selection of culturable representatives of the bacterial core microbiome to test the effects of fertilisation on growth in vitro.
DNA Extraction and Sequencing
2.2
Total DNA was extracted from ~200 mg of soil sample using the ZymoBIOMICS DNA Miniprep Kit (Zymo Research, USA) following the manufacturer's instructions. DNA concentration was determined after extraction and ~5–20 ng of DNA was used for amplification of the V4 region of 16S rRNA genes as described elsewhere (Fackelmann et al. 2021). In brief, a 291 bp fragment was amplified using the 515F (′5‐GTGCCAGCMGCCGCGGTAA‐3′) and 806R primers (5′‐GGACTACHVGGGTWTCTAAT‐3′) (Caporaso et al. 2011) following the earth microbiome protocol and recommendations for polymerase chain reaction (PCR) amplification. This first PCR included 1 μL template DNA, 5 μL AmpliTaq Gold 360 Master Mix (Applied Biosystems, Darmstadt, Germany), 1.5 μL TS primers, and 2.5 μL water to give a final volume of 10 μL. The amplification protocol included an initial denaturation of 10 min at 95°C, followed by 30 cycles with 95°C for 30 s, 58°C for 30 s and 72°C for 45 s, followed by a final elongation of 72°C for 7 min. In a second PCR, amplicons were barcoded. Primers for barcoding were tagged with universal adapters (CS1 and CS2, Standard BioTools, South San Francisco, USA) and 4 Ns were added to the forward primer for cluster identification during sequencing. PCR reactions contained 3 μL template amplicon from the unbarcoded sample, 4 μL individual barcodes and Illumina adapters (Fluidigm Access Array System for Illumina Sequencing Systems, Standard BioTools, South San Francisco, USA), and 3.0 μL ultrapure water for a total of the 20 μL reaction. The reaction times were identical to the first PCR, but only 10 cycles were carried out. All PCRs were performed on a SimpliAmp Thermal Cycler (Applied Biosystems, Darmstadt, Germany) with negative controls containing only the reagents included and subsequently sequenced.
Barcoded PCR products were purified to remove residual oligonucleotides using the NucleoMag NGS Clean‐up and Size Select Kit (Macherey‐Nagel, Düren, Germany) on a GeneTheatre (Analytik Jena, Jena, Germany) according to the manufacturer's guidelines and in‐house workflow and checked for expected amplicon length using capillary electrophoresis on a QIAxcel Advanced System (QIAGEN, Hilden, Germany). DNA concentrations of the barcoded samples were quantified using the QuantiFluor dsDNA System (Promega, Madison, USA) on a TECAN Infinite F200 PRO plate reader (Tecan, Männedorf, Switzerland). Samples were normalised to include 60 ng of each indexed amplicon in the final library. For sequencing, 8 pM of the libraries were loaded onto a MiSeq flow cell and spiked with PhiX sequencing control V3 at 5% (Illumina MiSeq Reagent Kit V2). Paired‐end sequencing was performed over 2 × 251 cycles in an Illumina MiSeq (Illumina, San Diego, USA) following the manufacturer's recommendations.
Analysis of the Bacterial Soil Microbiome
2.3
Raw reads were analysed in QIIME 2 v2022.8 (Bolyen et al. 2019). DADA2 (Callahan et al. 2016) was used for quality filtering and assembly of reads into amplicon sequence variants (ASVs) and SILVA v138.2 was used as a taxonomic reference database. ASVs, taxonomy, metadata and phylogeny were imported into R package phyloseq v1.40.0 (McMurdie and Holmes 2013). ASVs identified in the blanks and controls were removed from samples to avoid false results and ASVs classified as mitochondria, chloroplasts and archaea were filtered out. We applied an additional filter to remove rare ASVs with fewer than 10 reads across the entire dataset. The raw reads were submitted to the NCBI database (BioProject: PRJNA1188490). Samples analysed in this study are the subset consisting of the soil samples and associated accession numbers are provided in Table S1.
All downstream analyses and visualisation were performed in R v4.2.2 (R Core Team 2022). To reflect the inherent spatial variation of bacterial communities within the experimental sites as well as to avoid observations based on singularities, we analysed individual soil samples nested within sites. The impact of organic fertilisation on soil microbial communities was investigated by analysing the distribution of taxa among fertilisation regimes. Absolute numbers of taxa unique to or shared by different experimental groups were extracted using the ‘get_vennlist’ function and were plotted with the R package VennDiagram v1.7.3 (Chen and Boutros 2011). Pielou's evenness indices of shared taxa were calculated for each fertilisation regime using the ‘diversity’ function of the R package VEGAN v2.6.4 (Dixon 2003). The effect of fertilisation regime on species evenness was assessed by fitting a generalised linear mixed model (GLMM) with a Gamma distribution (log link). To account for the hierarchical sampling design, treatment was included as a fixed effect, while site and sample (nested within sites) were specified as random intercepts. Core taxa were selected among shared soil microbial communities by applying cut‐off values for prevalence (present in at least 50% of all samples) and abundance (detection threshold > 0.01% relative abundance) using the ‘subset_taxa’ function of the phyloseq R package and only genera with valid taxonomic descriptions were retained. Differences in the relative abundances of core bacterial members were assessed using GLMMs with abundances transformed for beta regression and modelled with a beta distribution (log link), with samples nested within site as a random factor. Heatmaps clustering ASVs according to prevalence across all samples and relative abundance within samples were generated using the ‘heatmap’ function of the R package ComplexHeatmap v2.9.2 (Gu et al. 2016). Additionally, to identify potential fertilisation‐responsive bacterial biomarkers, we used the MaAsLin2 package v1.12.0 (Mallick et al. 2021) with the same model structure as described above (samples nested within sites). Core bacterial taxa were agglomerated prior to analysis, and the data were normalised using total sum scaling and log‐transformation. PICRUSt2 v2.5.2 (Douglas et al. 2020) was used to predict the functional potential of the bacterial biomarkers at the study sites. This software package allows predictions on the functional potential of a bacterial community based on abundance profiles of marker gene sequences such as 16S rRNA gene amplicons. The obtained KEGG orthologs were used to compute functional diversity and its response to various fertilisation regimes. To test the effect of fertilisation‐induced changes in the bacterial community on key metabolic pathways, a GLMM using a Gamma distribution (log link) was applied, again with treatment as a main effect and sample nested within site as a random factor. Finally, correlation between the bacterial biomarkers and functional diversity was tested by Spearman correlation using the ‘cor_test()’ function in base R, which accounts for the difference in the data distribution for various biomarker bacteria. All R code used for statistical analysis and visualisation is publicly available on GitHub (https://github.com/jk00ANI/Impact‐of‐Fertilization‐on‐Grassland‐Core‐Soil‐Microbiota‐Abundance‐in‐the‐Field‐and‐Growth‐in‐vitro).
Bacterial Strains and Cultivation of Biomarker Bacteria
2.4
Since 16S rRNA gene analysis did not provide species‐level resolution, representative species from the identified core genera available in public strain repositories were selected. For further experiments, bacteria were selected based on the following criteria: (i) Culturable type strain, (ii) Biosafety level 1 (iii) isolated from soil if possible. This led to an initial selection of 7 potential biomarker strains (Table S5), which were purchased from public strain repositories and recovered using media and culture conditions recommended by the supplier. Growth of candidate biomarker strains was evaluated on agar plates with seven different media at 30° and 37°C, totaling 14 conditions. Media tested included the following six widely used complex media reported to support the cultivation of a wide range of bacteria: nutrient agar (NA), brain heart infusion (BHI), Reasoner's 2A (R2A), Luria Bertani (LB), tryptic soy agar (TSA) and 2xTY. All media were prepared according to the formulations and protocols provided by the German Collection of Microorganisms and Cell Cultures (DSMZ; https://www.dsmz.de/collection/catalogue/microorganisms/culture‐technology/list‐of‐media‐for‐microorganisms). In addition, growth was tested on a custom‐made soil medium (SM) that was formulated to mimic the nutritional conditions of the study sites (Supporting Information).
Growth of biomarker candidates was tested in liquid media of R2A, NB and SM in microtiter plates using a BioTek Synergy H1M (Supporting Information). The effect of PS on the growth of selected culturable biomarker bacteria was investigated in shake flasks. R2A medium was prepared and PS was added to a final concentration of 2% (v/v), which is an estimation of the amount of PS applied in a single fertilization. To exclude contamination by bacteria present in PS, solids were removed by centrifugation and the remaining supernatant was filter‐sterilized twice prior to addition to R2A medium. Biomarker bacteria were grown to stationary phase in 5 mL of R2A medium in glass tubes. Bacteria were then washed with sterile saline (0.9% (w/v) NaCl in H_2_O) and used to inoculate 50 mL of R2A medium with or without 2% (v/v) PS to a starting OD_600_ of 0.1. The cultures were incubated at 30°C and growth was monitored by measuring OD_600_ until the stationary growth phase was reached. Growth rates and carrying capacity for individual cultures were calculated using the R package Growthcurver v0.3.1 that fits data to a logistic equation (Sprouffske and Wagner 2016). Growth curves were plotted and statistical analysis of growth rates and carrying capacity was performed using GraphPad Prism (Mavrevski et al. 2018).
Results
3
Effects of Different Fertilisation Regimes on Diversity of Soil Bacteria in Grassland Ecosystems
3.1
To explore the impact of organic fertilisation on the diversity of bacterial communities in the soil of the grassland ecosystem, we collected soil samples from study sites managed with different fertilisation regimes: pig slurry (PS), cow manure (CM), biogas digestate (BD) and control soil (CS). Following DNA isolation and 16S rRNA gene amplicon sequencing, we obtained a total of 2,303,276 reads that passed the quality criteria. Based on 100% sequence identity, 5769 distinct ASVs were identified, of which 992 were shared among all fertilisation regimes. These 992 ASVs represented 84.7% of all reads in the dataset. 112–292 ASVs were found to be shared by at least two fertilisation regimes, and 605–925 ASVs were unique to a specific regime (Figure 2A). This suggested an uneven distribution of unique and shared taxa among the different fertilisation regimes and a strong representation of the shared bacterial core microbiota.
Differences in microbial community composition of grassland soil fertilised with pig slurry (PS), biogas digestate (BD), or cow manure (CM) or soil samples from minimally fertilised control sites (CS). (A) Venn diagram of unique and shared bacterial taxa found in soils fertilised with different organic regimes. (B) Pielou's Evenness indices were calculated using the abundance of the 992 shared ASVs within the different fertilisation regimes. Effects of fertilisation regimes on Pielou's Evenness were tested using generalised linear mixed effect models.
To analyse the variation in abundance of the 992 shared ASVs, Pielou's Evenness indices (J) were compared (Figure 2B) and an overall high evenness in all treatment groups with no significant differences was observed (Table S2).
We next sought to identify the most abundant, known, and culturable representatives of the bacterial core microbiota. To this end, the 992 ASVs of the soil core bacteria were filtered for those that were present in at least 50% of all samples with a relative abundance of at least 0.01%. Moreover, ASVs without valid taxonomic identification at the genus level (unknown or Candidatus species/genera) as well as those classified as unculturable were excluded. This resulted in a total of 33 genera representing the validly described core soil bacteria of our data set. These 33 genera showed characteristic patterns of abundance in each of the fertilisation regimes and the differences in abundance patterns were most pronounced between CS and PS‐treated soil (Figure 3A).
Abundance and functionality of the bacterial core soil microbiota of grasslands under different fertilisation regimes. (A) Community composition of the bacterial soil core microbiota of grasslands fertilised with pig slurry (PS), biogas digestate (BD), or cow manure (CM) or soil samples from minimally fertilised control sites (CS). (B) Comparison of the relative abundance of KEGG orthologs for carbohydrate, methane, nitrogen and sulfur metabolism at study sites of the four fertilisation regimes. Effects of fertilisation regimes on relative abundance of KEGG orthologs were modelled using generalised linear mixed models (: p < 0.05; **: p < 0.01; **: p < 0.001).
To assess if these changes in abundance patterns of the bacterial core microbiota also may have consequences on the functional level, we predicted the KEGG orthologs for carbohydrate, methane, nitrogen and sulfur metabolism associated with ASVs of the 33 core genera across all samples and fertilisation regimes using PICRUSt2 (Douglas et al. 2020). This suggested a substantial reduction in KEGG orthologs of all four functional categories by all fertilisation regimes compared to CS (Figure 3B, Table S3). The predicted reduction in functional orthologs for carbohydrate metabolism was significant for all three fertilisation regimes (BD: β = 0.311, p < 0.001; CS: β = −0.230, p < 0.05; PS: β = 0.317, p < 0.001). KEGG orthologs for nitrogen metabolism were significantly reduced in BD (β = −0.218, p < 0.05) and PS (β = −0.230, p < 0.01) samples. Similarly, a significant reduction in KEGG orthologs for sulfur metabolism was predicted for BD (β = −0.195, p < 0.05) and PS (β = −0.209, p < 0.05). KEGG orthologs for methane metabolism were also reduced by all treatments but this effect only was marginally significant in BD samples (β = 0.158, p = 0.052).
Microbial Biomarkers of Soil Fertilisation Status
3.2
Based on the characteristic patterns of abundance of the bacterial core microbiota, we hypothesized that some of these genera may be potential biomarkers of soil fertilisation status. We thus further analysed the genera of the bacterial core microbiota to identify taxa that are representative of the observed differences between fertilisation regimes. To narrow down the list of potential biomarker bacteria, we sorted the 33 known genera according to prevalence at different levels of relative abundance (Figure 4A). Filtering for genera with a prevalence of at least 75% in all samples at a detection threshold of at least 0.05% relative abundance provided a total of 12 known genera, which were used for further analysis.
(A) Abundance and prevalence of known bacteria genera across fertilisation regimes. (B) Potential microbial biomarkers in soil of grassland ecosystems of the Swabian Alb indicative of untreated CS soil or fertilisation with BD, CM or PS. Biomarkers were identified by comparing relative abundance of the 12 genera shown in (A) computing effect size using MaAsLin2 (: p < 0.05; **: p < 0.01; **: p < 0.001).
To identify those bacterial genera that may serve as biomarkers for specific fertilisation regimes, we applied MaAsLin2, a generalised linear mixed model. Estimation of effect size, the generalised linear models of MaAsLin2 allow identification of taxa that most likely explain differences between fertilisation regimes considering their abundance. Significant association with any of the regimes was observed for 7 genera (Figure 4B). The genera Bacillus, Reyranella, and Bradyrhizobium are characteristic of CS sites. Gaiella was associated with BD, CM, and PS, and Nocardioides, Solirubrobacter, and Pseudocardia are associated with PS‐fertilised soil.
We then compared the relative abundance of the 7 biomarker genera in samples of all fertilisation regimes using a generalised linear mixed model with a beta distribution (log link) (Figure 5, Table S4). CS sites show predominance of Bacillus (CS vs. BD: β = −0.761, p < 0.01; CS vs. CM: β = −1.006, p < 0.001; CS vs. PS: β = −1.38, p < 0.001), Bradyrhizobium (CS vs. BD: β = −0.338, p < 0.01) and Reyranella (CS vs. BD: β = −0.252, p < 0.001; CS vs. CM: β = −0.203, p < 0.001; CS vs. PS: β = −0.137, p < 0.05). Genera that were predominant on fertilised sites were Gaiella (CS vs. BD: β = 0.396, p < 0.001; CS vs. CM: β ~= 0.386, p < 0.001; CS vs. PS: β = 0.671, p < 0.001), Nocardioides (CS vs. PS: β = 0.315, p < 0.01), Pseudonocardia (CS vs. PS: β = 0.525, p < 0.001), and Solirubrobacter (CS vs. PS: β = 0.229, p < 0.01). This highlights that PS fertilisation has the most profound impact on the abundance of the identified biomarker bacteria.
Response of potential bacterial biomarker genera to different fertilisation regimes (PS, BD, CM, and CS) applied to grassland sites in the Swabian Alb. Abundance of biomarker bacteria was transformed prior to analysis and fitted using generalised linear mixed models (GLMMs) with a beta distribution (: p < 0.05; **: p < 0.01; **: p < 0.001).
Culturable Representatives of the Bacterial Core Soil Microbiota
3.3
To study the effect of fertilisation on the growth of representative members of core soil bacteria in the lab, we searched public microbial culture collections for type strains of the 7 potential biomarker genera identified by MaAsLin2. For genera with several type strains available, preference was given to strains isolated from soil and, when possible, widely used model strains. We found type strains for all genera (Table S5) but failed to obtain Reyranella soli NBRC 108950^T^ even after repeated efforts. Although Gaiella occulta CECT 7815^T^ was successfully grown in the lab on its recommended medium (data not shown), this strain was excluded from further experiments due to extremely slow growth. For the remaining five potential biomarker genera, type strains isolated from soil or root nodules were successfully grown in the lab on agar plates of the media recommended by the provider, and these strains were used for further analysis.
To identify conditions of cultivation that support the growth of a maximum number of biomarkers, Bacillus subtilis DSM10^T^, Bradyrhizobium japonicum DSM 30131^T^, Nocardioides albus DSM 43109^T^, Pseudonocardia antarctica DSM 44749^T^, and Solirubrobacter pauli DSM 14954^T^ were tested on agar plates of six different standard media plus a custom‐made soil medium (SM). Nutrient Agar (NA), Reasoner's 2A (R2A) agar, and SM agar supported the growth of all type strains tested (Table S5) and were thus used to test the growth of all strains in liquid culture. P. antarctica showed good growth but formed aggregates in all media (data not shown), which prevented reliable measurement of OD_600_. Aiming for a rapid comparative assessment of growth characteristics of core soil representatives in response to fertilisation, this strain was also excluded from further experiments.
The remaining four potential biomarker bacteria were compared for growth on R2A, nutrient broth (NB) and SM in microtiter plates (Figure S1). This revealed that all three media supported the growth of the selected bacteria to some degree but with strong differences in growth rates and final optical densities between strains and across media. Moreover, considerable variability between individual experiments was noted resulting in high standard deviations of the mean. This was mainly attributed to strong differences in lag phase and was particularly evident for N. albus DSM 43109^T^ and S. pauli DSM 14954^T^ on SM and NB.
Based on these results, R2A medium was the medium that gave the most consistent results for all four strains and selected for a comparative analysis of the effect of PS on the growth of the four selected core soil bacteria in shake flasks (Figure 6). Interestingly, the effects of PS on growth in vitro mirrored abundance patterns observed in the field samples. In line with a reduced abundance in PS‐fertilised soil samples (Figure 5), growth rates and carrying capacity of B. japonicum were reduced in the presence of PS and this effect was significant for carrying capacity (Figure 6). Similarly, growth rates of B. subtilis were significantly reduced by PS but carrying capacity was, however, increased. Conversely, growth rates and carrying capacity of N. albus and S. pauli in R2A were significantly increased in the presence of PS (Figure 6) matching their increased abundances in PS‐fertilised soil samples (Figure 5).
Effect of pig slurry on growth of culturable soil representative bacteria in vitro. All four biomarker bacteria were grown on R2A medium with (filled circles; +PS) or without (open circles; −PS) 2% (v/v) pig slurry. Growth was monitored by measuring OD600 at the indicated timepoints. Growth rates (GR) and carrying capacity (CC) for individual cultures were calculated by fitting OD600 values to a logistic equation and are plotted below growth curves. All values are mean ± standard deviation of n = 3 independent cultures per strain. Statistical analysis was performed by Student's t‐test on GR and CC calculated for individual cultures.
To finally test if there are differences in the functional capacity of the members of the genera Bacillus, Bradyrhizobium, Nocardioides, and Solirubrobacter depending on the fertilisation regime we again performed PICRUSt2 analyses. Using KEGG orthologs inferred from ASVs we calculated the functional diversity (Shannon indices) in each sample for the four genera separately. Analysing functional diversity vs. relative abundance (Figure 7; Table S6) revealed a significant positive correlation for the genera Bacillus (ρ = 0.73, p = 9.7 × 10^−17^) and Bradyrhizobium (ρ = 0.4, p = 8.2 × 10^−5^) indicating that higher abundance is associated with higher (predicted) functional diversity. Conversely, a significant negative correlation was observed for Nocardioides (ρ = −0.28, p = 0.0083) and Solirubrobacter (ρ = −0.21, p = 0.046) suggesting that the higher the abundance of these genera at a fertilised site is, the lower is their (predicted) functional diversity. Moreover, Bacillus and Bradyrhizobium appeared to be less abundant on PS‐fertilised sites and these communities also were predicted to have lower functional diversity. By contrast, Nocardioides and Solirubrobacter showed higher abundance on CS‐fertilised study sites and these bacteria were predicted to have a higher functional diversity.
Spearman correlation between the functional diversity (Shannon index) and abundance of the potential bacterial biomarkers of fertilisation. Functional diversity was calculated using PICRUSt2 based on KEGG orthologs in genomes of the database belonging to the ASVs at each study site.
Discussion
4
In this study, we examined the impact of organic fertilisation regimes on the bacterial soil microbiome of grassland ecosystems in the Swabian Alb. Our aim was to identify culturable representatives of the bacterial core microbiota as biomarkers of soil fertilisation status, which could be used to investigate the effects of fertilisation and other anthropogenic factors on microbial growth in vitro. Using high‐throughput 16S rRNA gene amplicon sequencing, we identified 5769 distinct ASVs in a total of 2,303,276 reads. These numbers are in accordance with other studies on the soil microbiome. A recent study assessing the effect of chemical fertilisation on soil microbial diversity in a controlled setting with pot‐grown wheat plants found 10,298 ASVs in 5.3 million reads of 286 samples (Reid et al. 2024). A large survey on microbial diversity of soil of different vegetation types across Europe found on average 8000–8300 bacterial OTUs in soil samples of extensively and intensively managed grasslands, respectively (Labouyrie et al. 2023).
Approx. 85% of sequence reads in our data set belong to taxa shared by all fertilisation regimes. Our sequencing data suggested that organic fertilisation had a profound impact on the structure of the core bacterial soil microbiome and these effects were most pronounced in study sites fertilised with PS. Filtering shared ASVs for highly abundant and prevalent taxa with validly described and culturable representatives identified 12 potential biomarker genera, which were further analysed using MaAsLin2, a common method to identify biomarkers indicative of specific conditions in microbiome data (Mallick et al. 2021). Additionally, the functionality of these potential biomarker genera was inferred using PICRUSt2‐based predictions. We observed asignificant reduction in KEGG orthologs related to carbohydrate, nitrogen, and sulfur metabolisms indicating that fertilisation regime may disrupt key soil microbial function and consequently influence nutrient cycling. Soil microbes play crucial roles in breaking down organic matter, fixing nitrogen and processing carbon storage with important consequences for ecosystem functions such as soil fertility and plant productivity (Fierer 2017; Delgado‐Baquerizo et al. 2020; Yadav et al. 2021). While our study provides important indications for functional change under organic fertilisation, further research is needed to explore the mechanism, by which a specific treatment influences soil microbial function.
Type strain of potential biomarker genera were obtained from culture collections and, based on their ability to grow on standard media, four species ( B. subtilis , B. japonicum , N. albus , and S. pauli ) were selected to investigate the effect of pig slurry on growth in vitro. Results of growth experiments with these four biomarkers reflected their abundance in the field and are mostly in line with their classification according to their hypothesized life strategies. According to the oligotroph‐copiotroph concept, oligotrophic bacteria that grow slowly under apparent optimal conditions are more prevalent in environments with low availability of nutrients whereas copiotrophs are fast‐growing organisms common in high‐nutrient environments (Koch 2001; Ho et al. 2017). These differences in life strategy may also help explain the relative growth rate and hence population size of bacterial groups in a given habitat. Solirubrobacter and Nocardioides showed higher abundance in PS‐fertilised study sites. Additionally, growth rates and final biomass of N. albus and S. pauli were increased in the presence of PS. Both genera belong to the phylum Actinomycetota, which contains mostly copiotrophic organisms (Stone et al. 2023). Thus, increased growth rates in the presence of higher levels of nutrients may, at least in part, explain the higher abundance of these organisms on PS‐fertilised soil.
According to the MaAsLin2 analysis, Bacillus is a biomarker for CS samples, which were obtained from minimally fertilised sites, and the abundance of ASVs belonging to this genus was significantly reduced in the soil of PS‐, CM‐ and BD‐treated sites. This contradicts the classification of the genus Bacillus as copiotrophs (Stone et al. 2023). Recently, Xu et al. examined the factors that affect the abundance of Bacillus subtilis group species in soil and found higher abundance in grassland soil compared to forest soil (Xu et al. 2024). At the same time, forest soils had higher organic content and less acidic pH and both factors correlated with abundance patterns. Nutrient availability is one of the major drivers shaping the soil microbiome (Fierer 2017) and organic fertilisers are a rich source of nutrients including organic carbon, nitrogen and phosphorous (Marinari et al. 2000; Plaza et al. 2007). Thus, the introduction of organic content by fertilisation might explain the reduced abundance of the genus Bacillus under all three fertilisation regimes in our study. Of note, B. subtilis DSM10 displayed reduced growth rates but higher final OD_600_ when grown in vitro in the presence of PS. While reduced growth rates would actually be in line with reduced abundance in conditions with higher nutrient availability, accumulation to higher biomass somehow contradicts the observations from the field. However, higher abundance of the genus Bacillus in low nutrient grassland soil was found to occur together with other genera of the Bacillota phylum in strongly interconnected modules (Xu et al. 2024), which may indicate cooperation. Such cooperative interactions as well as potential antagonistic interactions in the field, in which one member of the consortium inhibits another, cannot be modelled by single strain cultivations in vitro.
As members of the Alphaproteobacteria, the genus Bradyrhizobium belongs to the oligotrophic organisms associated with low‐nutrient environments (Stone et al. 2023). In line with this classification, Bradyrhizobium was suggested by MaAsLin2 as another biomarker genus indicative of untreated soil samples and the abundance of ASVs was significantly reduced in BD‐fertilised sites. Similar to our results, a lower abundance of bacterial nitrogen‐fixers was observed in intensively vs. extensively used grasslands in a large survey of microbial diversity in soils across Europe (Labouyrie et al. 2023). B. japonicum is a member of the Rhizobiaceae family of bacterial symbionts that are actively recruited by legume plants into specialised root nodules for nitrogen fixation (Hungria et al. 2015). Recruitment of nitrogen‐fixing bacteria by plants is mediated by root exudates that are regulated by environmental stimuli (Canarini et al. 2019; Trivedi et al. 2020), shape the composition of the soil microbiome (Seitz et al. 2022), and may have a profound impact on the nitrogen cycle in soil (Coskun et al. 2017). Thus, the reduced abundance of ASVs belonging to the genus Bradyrhizobium in fertilised soil could be explained by a down‐regulation of exudates by plants in response to nitrogen influx in reduced numbers of nitrogen‐fixing bacteria. Our in vitro growth experiments revealed that PS strongly inhibited the growth of B. japonicum . It is thus possible that the reduction of Bradyrhizobium spp. in fertilised soil may be a direct consequence of growth inhibition by organic fertilisers.
In summary, our study provides culture‐independent and ‐dependent evidence of the impact of organic fertilisation on the structure of bacterial soil microbiomes. Using a large 16S rRNA gene amplicon data set obtained from field samples, we were able to show that a large portion of the observed taxa is shared across all sampling sites and define the core microbiota of known bacterial genera. This bacterial core microbiota responded to different organic fertilisation regimes by characteristic changes in abundance patterns. Building on core‐microbiome and differential abundance, MaAsLin2 analysis allowed identification of biomarker genera that show characteristic differences in abundance on PS‐fertilised and CS sites. The effects of PS on the growth of culturable representatives of these biomarker genera are in line with their observed changes in abundance in field samples. Thus, further in vitro experiments may provide a tool to assess the impact of individual parameters of organic fertilisation or other environmental factors on members of the bacterial core soil microbiota. Given the high genetic variability even within one species and, even more so, on the genus level, results obtained with type strains in in vitro growth experiments are somewhat speculative and only a first indication of the effects of organic fertilisation on individual components of the bacterial core microbiome. Consequently, the presented in vitro systems and the findings need to be validated in further experiments with for example, primary soil isolates and under more natural conditions reflecting temperature fluctuations.
Author Contributions
Rostand R. Chamedjeu: investigation, formal analysis, writing – original draft, writing – review and editing. Kunal Jani: methodology, investigation, formal analysis, validation, writing – review and editing, data curation. Karoline Jetter: investigation, formal analysis, writing – review and editing. Kerstin Wilhelm: investigation, methodology, writing – review and editing. Patrick Schäfer: conceptualization, funding acquisition, writing – review and editing, supervision, resources. Lena Wilfert: conceptualization, funding acquisition, writing – review and editing, formal analysis, supervision, resources. Simone Sommer: conceptualization, funding acquisition, formal analysis, writing – review and editing, supervision, resources. Christian U. Riedel: conceptualization, funding acquisition, validation, writing – original draft, writing – review and editing, supervision, resources.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Data S1: emi470235‐sup‐0001‐supinfo.pdf.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Babin, D. , C. Leoni , A. L. Neal , A. Sessitsch , and K. Smalla . 2021. “Editorial to the Thematic Topic Towards a More Sustainable Agriculture Through Managing Soil Microbiomes.” FEMS Microbiology Ecology 97: fiab 094.34263312 10.1093/femsec/fiab 094 · doi ↗ · pubmed ↗
- 2Berg, G. , D. Rybakova , D. Fischer , et al. 2020. “Microbiome Definition Re‐Visited: Old Concepts and New Challenges.” Microbiome 8: 103.32605663 10.1186/s 40168-020-00875-0PMC 7329523 · doi ↗ · pubmed ↗
- 3Bissett, A. , A. E. Richardson , G. Baker , and P. H. Thrall . 2011. “Long‐Term Land Use Effects on Soil Microbial Community Structure and Function.” Applied Soil Ecology 51: 66–78.
- 4Bohan, D. A. , C. Vacher , A. Tamaddoni‐Nezhad , A. Raybould , A. J. Dumbrell , and G. Woodward . 2017. “Next‐Generation Global Biomonitoring: Large‐Scale, Automated Reconstruction of Ecological Networks.” Trends in Ecology & Evolution 32: 477–487.28359573 10.1016/j.tree.2017.03.001 · doi ↗ · pubmed ↗
- 5Bolyen, E. , J. R. Rideout , M. R. Dillon , et al. 2019. “Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2.” Nature Biotechnology 37: 852–857.10.1038/s 41587-019-0209-9PMC 701518031341288 · doi ↗ · pubmed ↗
- 6Callahan, B. J. , P. J. Mc Murdie , M. J. Rosen , A. W. Han , A. J. A. Johnson , and S. P. Holmes . 2016. “DADA 2: High‐Resolution Sample Inference From Illumina Amplicon Data.” Nature Methods 13: 581–583.27214047 10.1038/nmeth.3869 PMC 4927377 · doi ↗ · pubmed ↗
- 7Canarini, A. , C. Kaiser , A. Merchant , A. Richter , and W. Wanek . 2019. “Root Exudation of Primary Metabolites: Mechanisms and Their Roles in Plant Responses to Environmental Stimuli.” Frontiers in Plant Science 10: 157.30881364 10.3389/fpls.2019.00157 PMC 6407669 · doi ↗ · pubmed ↗
- 8Caporaso, J. G. , C. L. Lauber , W. A. Walters , et al. 2011. “Global Patterns of 16S r RNA Diversity at a Depth of Millions of Sequences per Sample.” Proceedings of the National Academy of Sciences 108: 4516–4522.10.1073/pnas.1000080107 PMC 306359920534432 · doi ↗ · pubmed ↗