Oxidative stress-induced stress granules: a central link to protein aggregation in neurodegenerative diseases
Neelam Younas, Inga Zerr

TL;DR
This paper explores how stress granules, under oxidative stress, contribute to protein aggregation in neurodegenerative diseases like Alzheimer's.
Contribution
The paper highlights oxidative stress as a central factor linking stress granules to pathological protein aggregation in neurodegeneration.
Findings
Stress granules can transition from protective to pathological under chronic stress.
Tau protein interacts with stress granules, promoting its aggregation in tauopathies.
Oxidative stress is identified as a key driver of this pathological shift.
Abstract
Intracellular aggregation of proteins such as Tau, TDP43, FUS, prion protein, and α-synuclein is a major hallmark of many major neurodegenerative diseases. Aberrant stress granules (SGs) are emerging as key contributors to the nucleation of toxic protein aggregates in these disorders. SGs are dynamic, membrane less cytoplasmic assemblies that form transiently through liquid–liquid phase separation (LLPS) of RNA binding proteins (RBPs) containing low complexity domains, together with stalled mRNAs, to help cells cope with stress. While physiological SGs facilitate cellular resilience to acute stress and undergo rapid disassembly, chronic or excessive stress leads to persistent SGs, driving pathological protein aggregation characteristic of age related neurodegeneration. The inherent reversible aggregation of RBPs crucial for cellular function paradoxically exposes them to misfolding…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
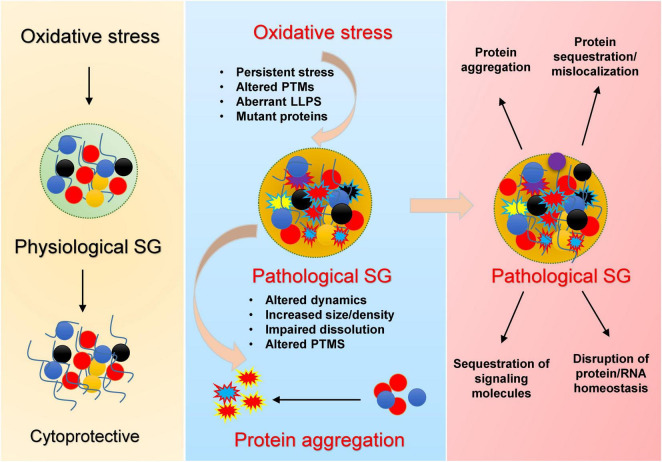 FIGURE 1
FIGURE 1| Protein | Stressor | Mutations | Model | PTM status | References |
|---|---|---|---|---|---|
| MAPT | Sodium arsenite, pathophysiological stress | P301L, P301S, G272V | Cell lines, mouse brain, human brain (endogenous) | Hyperphosphorylation, truncation, acetylation | |
| SFPQ | Sodium arsenite, pathophysiological stress | N533H, L534I | Cell lines, live cells, human brain | Oxidation, Phosphorylation | |
| TDP43 | Sodium arsenite, heat shock, sorbitol or paraquat, pathophysiological stress | G294A, A315T, Q331K, Q343R, G348C, M337V, R361S | Cell lines, primary glia, human brain or spinal cord | Oxidation, Phosphorylation, S-nitrosylation | |
| FUS | Sodium arsenite, traumatic injury or heat shock | R521C, R495X, H517Q, P525L | Mouse primary motor neuron, mouse spinal cord, drosophila brain or zebrafish spinal cord | Methylation, phosphorylation, acetylation | |
| PRNP | Sodium arsenite, pathophysiological stress | – | Cell lines, mouse brain, | Phosphorylation, glycosylation, truncation | |
| SNCA | Sodium arsenite, ↑OS interacts with mutations (H50Q, E46K), αS toxicity linked to P-bodies | H50Q, E46K, A53T, A30P | Cell lines, yeast, Drosophila, and human genetics | Phosphorylation (S129), nitration, sumoylation | |
| hnRNPA1 | Sodium arsenite, heat shock, or sorbitol, pathophysiological stress, OS induces stress granule pathology | D290A, P288A, D262V, *321Eext*6, *321Qext*6, and G304Nfs*3 | Cell lines, human fibroblast cells from ALS/FTD patients | Phosphorylation, PAR-ylation | |
| TIA1 | Sodium arsenite, pathophysiological stress | P362L | Cell lines, mouse brain, human brain | Oxidation, PAR-ylation | |
| ATXN2 | Sodium arsenite | Toxic polyQ expansions | Yeast cells, mouse (SCA2), cellular models | Oxidation, phosphorylation, LLPS interactions | |
| UBQLN2 | OS increases stress granules, aggregation | P497H, P497S, A488T | iPSC-motor neurons, KO mice | Phosphorylation, aggregation, altered LLPS |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRNA Research and Splicing · Genetic Neurodegenerative Diseases · Alzheimer's disease research and treatments
Introduction
1
A key hallmark of major neurodegenerative disorders—including Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), synucleinopathies and prion diseases—is the intracellular accumulation of aggregated proteins such as MAPT, TDP43, FUS, PRNP and SNCA (Wilson et al., 2023).
Although significant progress has been made in delineating the aggregation process, the exact cellular mechanisms that precipitate pathological aggregation remain inadequately resolved.
Newly identified elements of these aggregates are stress granules (SGs)–membrane less cytoplasmic assemblies of RNA-binding proteins and stalled RNA translational machinery–as crucibles of aggregation (Anderson and Kedersha, 2008; Fomicheva and Ross, 2021). Stress granule dynamics are conserved throughout eukaryotes and represent a universal cytoprotective mechanism against diverse environmental stresses (Kedersha et al., 2013) including oxidative stress, heat shock, osmotic stress, UV irradiation, and nutrient deprivation (Cabral et al., 2022; Glauninger et al., 2022; Kedersha et al., 1999). Under physiological conditions, SGs are transient, rapidly dissolving upon stress resolution. However, chronic or persistent SGs—often observed with aging or continuous stress exposure—may act as nucleation centers for disease-associated protein aggregation, thereby contributing to neurodegenerative pathology (Figure 1).
Mechanistic overview by which oxidative stress promotes pathological stress granule (SG) formation and protein aggregation. Under acute stress, SGs transiently assemble, sequester RNAs and proteins, and regulate translation, supporting cell survival during stress (left). Chronic/recurrent oxidative stress promotes persistent SGs, oxidation of key residues, abnormal PTMs, and LLPS transitions, especially in the presence of disease mutations (middle), leading to conversion of dynamic liquid-like SGs into dense, pathological condensates. These aberrant SGs display impaired dissolution, altered composition, and serve as nucleation sites for neurodegeneration-associated protein aggregates. Their persistence (right) drives protein misfolding and mislocalization, sequestration of signaling factors, disruption of protein/RNA homeostasis, and ultimately neuronal dysfunction.
Multiple lines of evidence provide a strong evidence that SGs in cells exposed to environmental stress play the role of seeds for pathogenic protein aggregation in neurodegenerative disorders. First, several proteins [MAPT (Maziuk et al., 2018; Vanderweyde et al., 2016; Younas et al., 2020), TDP43 (Yan et al., 2025), FUS, SNCA (Younas et al., 2023b) and PRNP (Younas et al., 2023b)] that mount up in cytoplasmic aggregates are also components of these SGs (Ash et al., 2021; LeBlang et al., 2020; Yan et al., 2025; Younas et al., 2023b). Second, long lifespan and high metabolism of neurons make them specifically vulnerable to successive stress episodes and to cycles of SG assembly-disassembly (Bishop et al., 2010). Third, mutations in the disease-linked proteins can disrupt normal dynamics of SGs (Lenzi et al., 2015; Mateju et al., 2017; Murakami et al., 2015). Additionally, mutations in other components of SGs e.g., hnRNPA1 (Clarke et al., 2021), TIA1 (Apicco et al., 2018; Baradaran-Heravi et al., 2020; Jiang et al., 2022), ATXN2 (Guevara-García et al., 2012; Kato et al., 2019), UBQLN2 (Peng et al., 2022; Yan et al., 2025) can also disrupt SG dynamics (Table 1). Fourth, SG dense cores act as hotspots for protein aggregation (Maziuk et al., 2018; Vanderweyde et al., 2016; Yan et al., 2025). Stress granules (SGs) have emerged as a critical missing link that integrates genetic, cellular, and pathological evidence, completing the puzzle of aberrant protein aggregation cascade in neurodegenerative diseases (Cui et al., 2024; Dudman and Qi, 2020; Gu et al., 2024; Yuan et al., 2025). A summary of key proteins, their associated stressors, reported mutations, models used for study, post-translational modifications (PTM), and supporting references are described in Table 1.
Oxidative stress-induced stress granules as key drivers of pathological aggregation
2
A central unresolved question in the field is whether specific stressors preferentially drive the conversion of physiological protein condensates into pathogenic aggregates in vivo, especially under prolonged or recurrent stress. A growing body of research points toward oxidative stress as a principal upstream trigger in this pathogenic cascade (Gu et al., 2024; Ratti et al., 2020). Recent work by Yan et al. (2025) demonstrates that the aggregation of TDP-43 within SGs not only requires high protein concentrations but also strictly depends on oxidative insults. Oxidative agents such as arsenite and paraquat oxidize cysteine residues, which is crucial for induction of TDP-43 demixing within condensates, finally converting it into pathological aggregates. By contrast, stress granules induced by non-oxidative agents (e.g., puromycin) fail to trigger aggregation—even at high TDP-43 levels and with impaired proteasomal or chaperone systems—unless oxidative stress is also present (Yan et al., 2025).
Similar mechanisms appear operative with Tau protein, the signature protein in AD and related tauopathies (Younas et al., 2020). Oxidative stress (induced by sodium arsenite) lead to the upregulation of TIA-1 (a classical marker of SGs) and phosphorylation of Tau in cellular models in both neuronal and non-neuronal cell lines, and human brain of AD cases (Alavi Naini and Soussi-Yanicostas, 2015; Younas et al., 2020). Tau phosphorylation–a hallmark of AD–promotes its phase separation and aggregation in vitro (Ambadipudi et al., 2017). Chronic or repeated oxidative stress, coupled with elevated levels of TIA-1 and phosphorylated Tau, may create a microenvironment within SGs that nucleates pathological aggregation. Thus, stress granules may represent the missing mechanistic link bridging oxidative damage, protein modification, and aggregate formation in neurodegenerative diseases (Figure 1).
Outstanding mechanistic questions remain, especially regarding which molecular species of Tau participate in SG formation. Are they distinct from the Tau species involved in the stabilization of microtubules? Previously it has been reported that a shift in localization of Tau toward somatodendritic compartments occurs to facilitate formation of SGs (Vanderweyde et al., 2016). However, it is unlikely that the same Tau species involved in microtubule stabilization are also responsible for stress granule formation, as this would compromise microtubules integrity during stress. It is more plausible that nuclear Tau species (Younas et al., 2023a) might be involved in the formation of SGs. Tau has six isoforms and many posttranslational modifications. Post-translational modifications also affect stress granules dynamics (Wang et al., 2023). Certain pathological Tau species—especially those that are hyperphosphorylated—show enhanced association with SGs, impacting SG dynamics and contributing to neurodegenerative mechanisms. This mechanistic link underscores the importance of isoform diversity and PTMs in how Tau modulates SG formation and pathological aggregation. Further research is needed to clarify this important issue. By focusing future research on the molecular interactions between oxidative stress, SG dynamics, and protein aggregation, this emerging concept has the potential to transform our strategies for early diagnosis, disease monitoring, and the development of novel treatments for Alzheimer’s disease and related tauopathies.
Chronic and recurrent oxidative stress in the context of mutant proteins—whether RNA binding or not—can drive the formation of pathological SGs (Table 1). Notably, mutations in SG-associated genes—including MAPT, TARDBP (TDP-43), FUS, ATXN2, TIA1, and HNRNPA1—have been shown to disrupt SG dynamics, impair disassembly, and enhance aggregation propensity, thereby contributing to neurodegenerative disease pathogenesis (Table 1).
This is also important to note that the vast majority of knowledge about oxidative stress and stress granule dynamics is derived from experimental exposure to artificial oxidants (e.g., sodium arsenite, H2O2) in cellular models. There is limited direct evidence demonstrating that physiologically relevant endogenous reactive oxygen species levels induce the same oxidative modifications and phase behaviors under disease conditions in vivo (Table 1). The conversion of protective, metastable condensates into pathogenic aggregates within stress granules is determined by a complex interplay of the protein involved, nature of oxidative insult, and the posttranslational landscape.
Taken together, these findings suggest that stress granules could represent a missing mechanistic link connecting oxidative damage, protein modification, and aggregate formation in neurodegenerative diseases (Figure 1). This mechanistic specificity underscores the potential for developing targeted, redox-oriented neuroprotective therapies aimed at modulating SG composition and stability under oxidative conditions. Advancement in this area will require deeper investigations into physiologically relevant stress profiles, diversity of isoforms and posttranslational modifications, and validation in robust in vivo models. These efforts hold promise for translating mechanistic insights into tangible clinical interventions that halt or reverse the aggregation process.
Conclusion
3
In conclusion, converging evidence positions stress granules as a mechanistic nexus where oxidative stress and altered protein homeostasis intersect to drive pathological aggregation in neurodegenerative disorders. The conversion of metastable, protective condensates into pathogenic aggregates appears to be highly contingent on the nature and duration of cellular insults—particularly oxidative stress—rather than on SG formation per se. This duality underscores the therapeutic potential of targeting stress granule dynamics and redox homeostasis, but also highlights the need for more granular understanding of the molecular players and contextual triggers involved. As our mechanistic insights deepen, SGs can offer a promising frontier for both biomarker discovery and intervention in proteinopathic neurodegeneration.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alavi Naini S. M. Soussi-Yanicostas N. (2015). Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid. Med. Cell Longev. 2015:151979. 10.1155/2015/151979 26576216 PMC 4630413 · doi ↗ · pubmed ↗
- 2Ambadipudi S. Biernat J. Riedel D. Mandelkow E. Zweckstetter M. (2017). Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 8:275. 10.1038/s 41467-017-00480-0 28819146 PMC 5561136 · doi ↗ · pubmed ↗
- 3Anderson P. Kedersha N. (2008). Stress granules: The Tao of RNA triage. Trends Biochem. Sci. 33 141–150. 10.1016/j.tibs.2007.12.003 18291657 · doi ↗ · pubmed ↗
- 4Apicco D. J. Ash P. E. A. Maziuk B. Le Blang C. Medalla M. Al Abdullatif A. (2018). Reducing the RNA binding protein TIA 1 protects against tau-mediated neurodegeneration in vivo. Nat. Neurosci. 21 72–80. 10.1038/s 41593-017-0022-z 29273772 PMC 5745051 · doi ↗ · pubmed ↗
- 5Arimoto-Matsuzaki K. Saito H. Takekawa M. (2016). TIA 1 oxidation inhibits stress granule assembly and sensitizes cells to stress-induced apoptosis. Nat. Commun. 7:10252. 10.1038/ncomms 10252 26738979 PMC 4729832 · doi ↗ · pubmed ↗
- 6Ash P. E. A. Lei S. Shattuck J. Boudeau S. Carlomagno Y. Medalla M. (2021). TIA 1 potentiates tau phase separation and promotes generation of toxic oligomeric tau. Proc. Natl. Acad. Sci. U. S. A. 118:e 2014188118. 10.1073/pnas.2014188118 33619090 PMC 7936275 · doi ↗ · pubmed ↗
- 7Baradaran-Heravi Y. Van Broeckhoven C. van der Zee J. (2020). Stress granule mediated protein aggregation and underlying gene defects in the FTD-ALS spectrum. Neurobiol. Dis. 134:104639. 10.1016/j.nbd.2019.104639 31626953 · doi ↗ · pubmed ↗
- 8Baron D. M. Kaushansky L. J. Ward C. L. Sama R. R. Chian R. J. Boggio K. J. (2013). Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol. Neurodegener. 8:30. 10.1186/1750-1326-8-30 24090136 PMC 3766239 · doi ↗ · pubmed ↗