Molecular characterization of Rhipicephalus microplus and Haemaphysalis bispinosa ticks from cattle across Thailand: Regional identification and evidence of different genetic sub-structures between mainland and peninsular populations
Danai Sangthong, Pradit Sangthong, Warin Rangubpit, Prapasiri Pongprayoon, Eukote Suwan, Kannika Wongpanit, Wissanuwat Chimnoi, Pacharathon Simking, Sinsamut Sae Ngow, Serge Morand, Roger W. Stich, Sathaporn Jittapalapong, Shawky Aboelhadid, Shawky Aboelhadid, Shawky Aboelhadid

TL;DR
This study characterizes tick species in Thailand, revealing genetic differences between mainland and southern populations, which could help improve tick control strategies.
Contribution
The study provides new insights into the genetic sub-structures of Rhipicephalus microplus ticks in Thailand and confirms the presence of H. bispinosa in the northeastern region.
Findings
Rhipicephalus microplus clades A and C were identified, with no evidence of clade B in Thailand.
H. bispinosa showed low genetic diversity, suggesting a bottleneck or founder effect.
Significant genetic differences were found between mainland and peninsular R. microplus populations.
Abstract
Phylogenetic and population genetic analyses were conducted on tick specimens collected from cattle in northern, northeastern, central, and southern regions of Thailand. Morphological identification indicated these ticks consisted of three species, Rhipicephalus microplus from all four regions, R. sanguineus from the northern and northeastern regions, and a Haemaphysalis species only collected from the northeastern region. Analysis of cytochrome c oxidase subunit I gene (COI) sequences identified R. microplus clades A and C, while clade B was not detected in this study. The same analysis indicated specimens morphologically identified as Haemaphysalis were H. bispinosa, confirming previous reports of their prevalence in northeastern Thailand. H. bispinosa showed low haplotype and nucleotide diversity, suggesting either a bottleneck or founder effect. Both R. microplus clades displayed…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
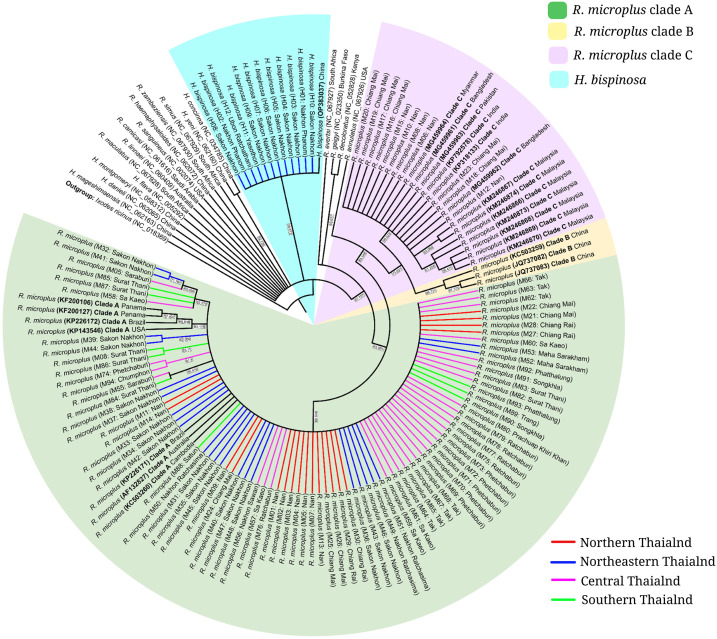 Fig 1
Fig 1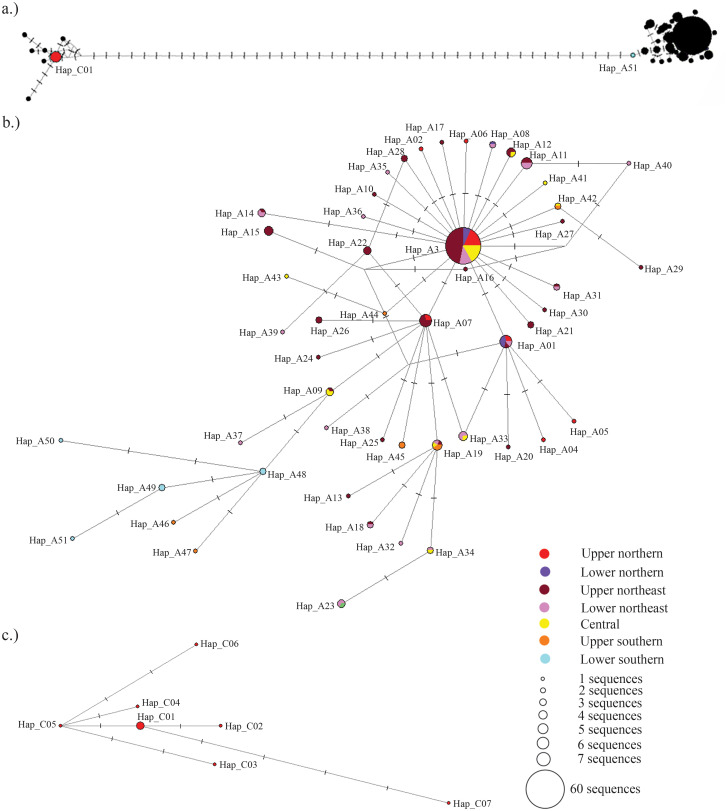 Fig 2
Fig 2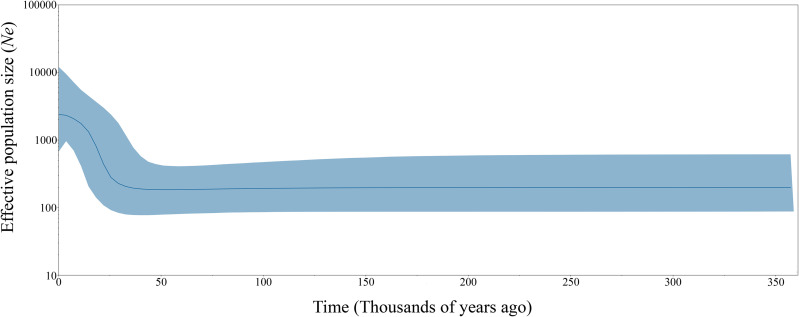 Fig 3
Fig 3- —http://dx.doi.org/10.13039/501100005621Kasetsart University Research and Development Institute
- —Kasetsart University Reinventing University Program 2021
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsVector-borne infectious diseases · Bartonella species infections research · Viral Infections and Vectors
Introduction
Ticks cause substantial economic losses to limited-resource farming communities, especially in tropical and subtropical regions where approximately 80% of the world’s cattle are raised [1,2]. Ixodid tick infestations cause direct damage due to feeding lesions and blood loss, and ticks transmit pathogens, including etiologic agents of four major arthropod-borne diseases of cattle, viz. anaplasmosis, cowdriosis, babesiosis, and theileriosis, which pose serious threats to both animal health and livestock productivity [3]. Thus, understanding local tick vector populations is fundamental to evaluation of tick-borne disease risks.
Rhipicephalus (Boophilus) microplus, commonly known as the “tropical cattle fever tick,” is arguably the most devastating hematophagous ectoparasite of cattle and buffaloes worldwide, in large part because it vectors agents of bovine babesiosis and anaplasmosis, which are considered the two most important vector-borne diseases of cattle worldwide [4]. Rhipicephalus microplus has been reported in Asia, including Southeast Asian countries such as Thailand [5–8]. Substantial losses in milk production are associated with tick infestation of dairy cows in Thailand because each engorged female tick is thought to be responsible for the loss of 8.9 ml of milk and 1 g of live weight gain [9]. Moreover, tick-borne diseases have been reported among cattle and buffaloes throughout Thailand [8,10–12]. Thus, tick distributions in Thailand are expected to be indicators of tick-borne disease outbreaks and their subsequent economic impact on livestock production. However, morphologic taxonomy of R. microplus is challenging, particularly due to the difficulty in morphological differentiation at the subspecies level [13]. Conversely, molecular characterization enables distinguishing closely related, morphologically identical taxa [14].
Acarine mitochondrial cytochrome c oxidase subunit I gene (COI) sequences from different parts of the world have become increasingly available in GenBank, allowing more robust phylogenetic comparisons [15]. To date, three distinct COI lineage assemblages are reported within the species R. microplus. Additionally, COI was identified as a suitable genetic marker for tick species identification, with five phylogenetic clades within a putative R. microplus complex: R. annulatus, R. australis, and three R. microplus s.l. clades (A, B and C) [16]. COI sequence analysis was useful in the classification and identification of these ticks [17,18]. However, genetic information on cattle ticks in Thailand remains limited and is primarily based on COI and 16S rDNA sequences from the northeastern region of the country. Analysis of COI sequences in this area revealed R. microplus clades A and C. The results obtained with COI and the highly conserved 16S rDNA sequences underscored the advantages of COI in resolving evolutionary relationships within R. microplus [19].
To obtain more comprehensive genetic information, this study employed COI sequences to address three objectives: [1] sampling and COI sequence analysis for identification of cattle tick species from all regions across Thailand, [2] conducting molecular analyses in greater depth at the population level, and [3] estimating the demographic history of tick populations in Thailand. Possible causes of these genetic characteristics were identified and future trends predicted for these populations. These findings are expected to be useful for guiding tick and tick-borne disease control strategies across Southeast Asia.
Materials and methods
Samples
Tick specimens were collected from cattle in 32 provinces across the four geographic regions of Thailand. Ticks were thoroughly rinsed with double-distilled water (ddH₂O), air-dried, and preserved in 70% ethanol at room temperature until further processing. Morphological identification was performed to differentiate the tick species, using previous publications as references [13,20–25]. All animal care and experimental procedures were approved by the Animal Experiment Committee of Kasetsart University, Thailand (Approval No. ACKU64-VTN-018), and were conducted in strict accordance with the Regulations on Animal Experiments at Kasetsart University. This field study involving vertebrate animals fully complied with these institutional regulations.
DNA extraction and PCR amplification
Ticks preserved in 70% ethanol were individually washed in ddH_2_O on a sterile plate before genomic DNA was isolated with the DNeasy Blood and Tissue Kit (Qiagen, Germany), and stored at −20 °C. Primers RmicCoI_parH1 (CTCAACTAATCATAAAGACATTGG) and RmicCoI_parL1 (TATAAACTTCAGGGTGGCCAA) were used to amplify the COI target sequence from each specimen. The optimized PCR consisted of 20 µL reactions containing 2 µL of 10X Taq Buffer, 1.25 mM MgCl_2_, 0.2 mM of each dNTP, 2 units Taq DNA polymerase (Thermo Fisher Scientific, Lithuania, EU), 0.250 µM of each primer, and 1 µL of each DNA template. The thermal cycler parameters included initial denaturation at 94 °C for 2 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 55 °C for 30 s, elongation at 72 °C for 30 s, and final elongation at 72 °C for 30 s. All PCR products were electrophoresed in 1% agarose gels. Amplicons were purified using the FavorPrep™ Gel/PCR Purification Kit (Favorgen, Taiwan) and submitted for Sanger DNA sequencing (ATGC Co.,Ltd., Thailand).
Phylogenetic analysis
COI sequences amplified from Rhipicephalus microplus (94 amplicons) and Haemaphysalis sp. (12 amplicons) were compared to 42 tick COI sequences retrieved from GenBank (S1 Table). These sequences were aligned, with Ixodes ricinus as the outgroup, using Muscle software [26]. A neighbor-joining (NJ) tree was reconstructed with 10^5^ bootstrap replications using the program PAUP v.4 (build 169) [27].
Population genetic analysis
Population genetics were based on the northern, northeastern, central, and southern geographic regions of Thailand. To determine the genetic structure of tick populations across Thailand, the number of haplotypes (h), haplotype diversity (Hd), nucleotide diversity (Pi), analysis of molecular variance (AMOVA), pairwise fixation index (FST), and gene flow (Nm) were analyzed using Arlequin v3.5.1.2 [28]. Genetic relationships between COI haplotypes of ticks in Thailand were accessed by reconstruction of Median Joining (MJ) Network using the program Network v10.2 (https://www.fluxus-engineering.com). Neutrality indices, including Tajima’s D, Fu’s Fs, and the divergence time parameter (τ), were determined using Arlequin v3.5.1.2. Historical demographic fluctuations were elucidated using a Bayesian Skyline Plot (BSP) in BEAST 2.6.0 [29]. A mutation rate of 3.54% per million years [30] was applied using the HKY substitution model, which was identified as the best-fitting model by jModelTest software [31]. An MCMC method with 100 million iterations under a relaxed exponential clock was performed. Effective sample size (ESS) and the construction of the BSP were determined using TRACER 1.6 software [32].
Results
Morphological survey of ticks on cattle in Thailand
A total of 341 tick specimens were manually classified, using morphological keys [13,20–25], into three species: Rhipicephalus microplus, R. sanguineus, and a member of the genus Haemaphysalis. Morphological keys used for species identification are summarized as follows:
Rhipicephalus (Boophilus) microplus: the basis capitulum was dorsally hexagonal, laterally produced, and equal in length to the mouthparts. The second palpal segments were not laterally produced but bore ridges on both the dorsal and ventral surfaces. This subgenus lacked festoons. Males possessed an adanal shield, an accessory adanal shield, and this species possessed a caudal process.Rhipicephalus sanguineus: the basis capitulum was dorsally hexagonal, laterally produced, and equal in length to the mouthparts. The second palpal segments were not laterally produced and lacked ridges on both the dorsal and ventral surfaces. This subgenus had festoons, and males possessed an adanal shield and an accessory adanal shield.Haemaphysalis: the second segment of the palp was laterally produced beyond the margin of the basis capitulum. The basis capitulum was rectangular in shape. This genus had festoons. Males did not possess an adanal shield, accessory adanal shield, or caudal process.
The prevalence of different species collected indicated that R. microplus was the most common species collected from cattle (78.59%), followed by members of the genus Haemaphysalis (16.72%) and R. sanguineus (4.69%). All three species were present in the northeastern region, while R. microplus and R. sanguineus were found in the northern region and R. microplus alone was collected in the central and southern regions (Table 1).
Table 1: Morphological identification of tick specimens collected from cattle in the four major geographic regions across Thailand. These specimens were morphologically identified with established taxonomic keys.
Molecular identification
Representative specimen samples were used for molecular analyses to confirm morphological identifications and to more specifically identify Haemaphysalis and R. microplus species and strains, respectively. The COI target sequence was not amplified from R. sanguineus. Details for each region, subregion and their respective provinces are presented in Table 2. Amplified COI sequences were compared to reference sequences and tree topology was clearly separated into two COI clades of Haemaphysalis and R. microplus. A monophyletic clade was formed between the samples identified morphologically as Haemaphysalis in this study and H. bispinosa (OP383037) from China, clustering separately from other Haemaphysalis spp. sequences (Fig 1). A monophyletic relationship was also observed between COI sequences of Thai ticks morphologically identified as R. microplus (Fig 1), which aligned most closely with Clades A and C. Clade A, which consisted of R. microplus sequences from all regions of Thailand, was the most common genetic lineage detected in this study. Clade B was not detected among sequences tested in this study. Clade C was only detected in samples collected from the northern region.
Table 2: Sampling locations of tick specimens used for COI sequence analysis. Numbers represent specimens of R. microplus and H. bispinosa collected throughout Thailand. Sources include original data from this study and previously published data [33].
Radial phylogenetic tree of tick COI sequences.The tree was constructed using PAUP software under the General Time Reversible (GTR) model, incorporating a Gamma-distributed rate of evolution and a proportion of invariant sites (GTR + G + I). Rate heterogeneity among sites was modeled using a Gamma distribution (shape parameter = 0.6550), with 49.60% of sites designated as invariant (pinvar = 0.4960). The tree depicts evolutionary relationships among tick species collected from various regions of Thailand. Bootstrap support values (based on 100,000 replicates) are shown at the nodes. Phylogenetic clades identified in this study are labeled as Rhipicephalus microplus clades A, B, and C, and Haemaphysalis bispinosa, represented by green, yellow, violet, and sky blue, respectively. Geographic origins of the tick samples—northern, northeastern, central, and southern Thailand—are indicated by branch colors: red, blue, pink, and green, respectively.*
Genetic diversity
Nucleotide diversity analysis of these sequences revealed a consistent pattern of low COI nucleotide diversity among both R. microplus and H. bispinosa across all four regions surveyed, while R. microplus and H. bispinosa respectively showed low and high haplotype diversity (Fig 2). H. bispinosa from the northeastern region exhibited a haplotype diversity of 0.439 and a nucleotide diversity of 0.00105. R. microplus clade A across Thailand showed haplotype diversity ranging from 0.670 to 0.956 and nucleotide diversity ranging from 0.00151 to 0.00531. R. microplus clade C exhibited a haplotype diversity of 0.818 and a nucleotide diversity of 0.00105. Haplotype analysis of 182 sequences revealed three haplotypes for H. bispinosa, 51 for R. microplus Clade A, and seven for R. microplus Clade C. Among the 51 haplotypes of R. microplus Clade A, Hap_A03 was most common, with a frequency of 37.74%, and was found in all except the southern region of Thailand. The other 50 haplotypes consisted of 15 shared haplotypes, all present in adjacent regions, and the remaining 35 were region-specific haplotypes (S2 Table).
Median joining network of R. microplus haplotypes from Thailand based on COI sequences, constructed using Network v10.2 software.Circle sizes represent the number of sequences belonging to each haplotype. Vertical bars on lines connecting haplotypes indicate the number of substitutions separating them. The geographic origins of each haplotype are indicated by different colors. The network relationship between R. microplus clades A and C is shown in (a). Connection details between haplotypes within clades A and C are presented in (b) and (c), respectively.
Genetic structure
Genetic differences between tick populations from different geographic regions in Thailand were analyzed based on various hypotheses of population structure (Table 3). The AMOVA results indicated genetic structure only when applying the two-region hypothesis, which divided the regions into the mainland (northern, northeastern and central) and peninsula (southern). Genetic variation between the mainland and peninsula accounted for 33.23% of the total variation and showed significant F-statistics (FCT = 0.332, p = 0.048). Genetic differences between the mainland and peninsular populations were reassessed using pairwise FST comparisons (Table 4), which ranged from 0.162 to 0.740, all of which were significant (p ≤ 0.05). In contrast, the pairwise FST values between populations within the mainland (northern, northeastern, and central) were non-significant, ranging from −0.019 to 0.076. However, the FST comparison between the mainland and peninsula showed a significant difference (FST = 0.350, p ≤ 0.05). The Nm values among the seven geographic regions ranged from 0.176 to infinity (Table 4). Nm values between the Lower Southern population and all other populations were below 1.00, while those for the Upper Southern population were higher, ranging from 1.227 to 5.212. In contrast, Nm values among the five mainland populations ranged from 6.091 to infinity.
Table 3: Hierarchical analysis of molecular variance (AMOVA) of 4 posited genetic structures. Bold values are statistically significant.
Table 4: Pairwise FST values (lower diagonal) and gene flow Nm (upper diagonal) between tick populations from different regions of Thailand. Values in the lower diagonal represent genetic differentiation (FST), while those in the upper diagonal indicate estimated gene flow (Nm) between populations. “Inf” denotes infinite gene flow. Statistically significant FST values are shown in bold.
Demographic history
Neutrality tests based on Tajima’s D and Fu’s Fs were conducted (Table 5). Negative values of Tajima’s D and Fu’s Fs were observed for H. bispinosa, which were collected only from northern Thailand, and from all regions for R. microplus clades A and C. Significant negative values of Tajima’s D test were detected in R. microplus clade A from the northeast and central regions, while significant and highly significant values of Fu’s Fs were detected in all populations of R. microplus. Pooled population analysis of R. microplus clade A, from all regions of Thailand, showed highly significant Tajima’s D and Fu’s Fs values of −2.20001 and −27.29267, respectively. A significantly negative value of Tajima’s D test indicated purifying selection or population expansion, while a significantly negative value of Fu’s Fs directly indicated population expansion. To assess population expansion, R. microplus sequences representing both clades were further analyzed using a skyline plot, with a substitution rate of 0.0354 per site per million years. BSP analysis revealed that the effective population size of R. microplus was stable over long periods of time and initiated a sudden expansion in the last 25,000 years (Fig 3).
Table 5: Summary of COI diversity of H. bispinosa and R. microplus clades A and C.
Demographic history of Rhipicephalus microplus in Thailand inferred using a Bayesian skyline plot based on COI sequences.The x-axis represents time in thousands of years before present (x 1000 years ago) and the y-axis represents the effective population size on a logarithmic scale. The blue solid line indicates the mean estimate, while the copper-blue shaded area represents the 95% highest probability density (HPD) interval.
Discussion
Thailand is an agricultural country with a diverse range of sectors. The livestock sector is a major industry with high market value, providing benefits to all levels of society [34,35]. However, tropical livestock production faces challenges, one of which is tick infestations [36]. At least 25 ixodid tick species have been identified in Thailand, belonging to the genera Haemaphysalis, Ixodes, Amblyomma, Dermacentor, Rhipicephalus and Nosomma [37]. In terms of threats to livestock health, particularly for cattle, only a few tick species have been reported, including R. haemaphysaloides, R. microplus, R. sanguineus and H. bispinosa [33,38,39].
Morphological identification results in this study revealed that R. microplus was prevalent throughout Thailand, suggesting R. microplus is a source of significant economic losses in livestock production due to its widespread distribution and ability to transmit pathogens such as Babesia bigemina, B. bovis and Anaplasma marginale [40]. Notably, R. microplus, R. sanguineus, and H. bispinosa, which are considered common tick species among livestock in Thailand, were identified among the specimens collected in this study. In contrast, R. haemaphysaloides was not detected in the current study, although a total of nine specimens of this species were previously reported from four of 18 provinces in the northeast region of Thailand [19]. Collectively, these reports support previous suggestions that R. haemaphysaloides was rarely found, only in the northeast region of Thailand [19], and suggests its limited presence in other regions. This distribution pattern aligns with reports of R. haemaphysaloides prevalence in Bhutan [41], China [42,43], and Sri Lanka [44]. Despite its limited distribution, R. haemaphysaloides remains a potential vector of disease agents and warrants continued surveillance [45].
Mitochondrial COI sequences were used to expand upon morphological identification of two species through molecular analysis, H. bispinosa and R. microplus. Other groups have used these sequences for specific identification of morphologically identical specimens, including a COI lineage restricted to R. microplus from China, which those authors suggested could be a cryptic species more closely related to R. annulatus [16]. In the current study, phylogenetic tree topology revealed a monophyletic clade of 12 Haemaphysalis sequences, which aligned most closely with the H. bispinosa reference sequence (OP383037). Taxonomic classification beyond the genus Haemaphysalis in Thailand, based on morphological characteristics, has been challenging. As a result, several authors reported such samples as “Haemaphysalis sp.” [46] or “Haemaphysalis spp.” [47–49]. The COI analysis performed in this study, incorporating a novel pair of primers and PCR protocol, overcame the limitations of morphological identification of Haemaphysalis spp., indicating these 12 Haemaphysalis sequences were H. bispinosa and the establishment of this species in northeastern Thailand.
The phylogenetic tree demonstrated COI-based identification of R. microplus aligned with morphological findings while offering enhanced resolution to the subspecies level. All 170 R. microplus sequences analyzed in this study were distinctly grouped into two clades, 159 sequences in clade A and 11 in clade C. R. microplus clade A was the most common across the four geographic regions sampled in Thailand, corroborating previous reports of its high prevalence in specific surveillance areas, such as the northeast region [19]. R. microplus clade A has been reported as the dominant and most widely distributed clade across subtropical and tropical regions worldwide [6,50–53]. Clade C, the other R. microplus lineage detected in this study, was also less frequent than clade A in other areas, including Pakistan, Myanmar, Malaysia, Bangladesh, India and Thailand [54–56]. Previously, 91 and 25 specimens of R. microplus clades A and C, respectively, were reported in studies focused on the northeastern region of Thailand [19]. However, another study in the same region detected only R. microplus clade A among the 79 R. microplus specimens examined [33]. In the present study, R. microplus clade C was found only in the northern region. The distribution of R. microplus clade C in Thailand, as well as in sub-regions of other countries where clade C was identified, warrants further investigation.
R. microplus clade B, which some authorities consider a cryptic species indigenous to Southern China and Northern India, was not detected in the current study and has not been reported in Thailand [19,33]. Based on this detailed investigation into the genetic lineages of R. microplus in Thailand, clade A is most likely the primary contributor to tick-borne disease outbreaks and the associated economic losses in this country. The less frequent clade C has an uncertain prevalence in Thailand, and its impact on livestock remains unclear. However, previously reported occurrences of clades B and C suggested they could potentially pose a threat in Thailand. Clade B has been reported in several states in northern India (Rajasthan, Punjab, Uttar Pradesh, Bihar, and West Bengal states) and southern China, while clade C has been widely reported in Pakistan, India, Bangladesh, Myanmar, and Malaysia [56]. These areas have continuous live cattle trade with Thailand, which likely facilitates the spread of ticks from all genetic lineages. Therefore, although clade B is currently undetectable and clade C remains less common, they could become more important due to the trade flows of live cattle across South and Southeast Asia, because Thailand imports live cattle from the west (India, Bangladesh and Myanmar) and exports them to several regions, including to the north (China and Laos), east (Cambodia and Vietnam) and south (Malaysia) [57]. Thus, trade routes could facilitate the spread of clades B and C that originate in other countries along Thailand’s western trade routes.
Haplotype analysis revealed clear distinctions between H. bispinosa and R. microplus, as well as between R. microplus lineages A and C, without haplotypes shared between clades or species. The absence of shared haplotypes among ticks in this study indicated a lack of gene flow, suggesting that they belong to distinct reproductive groups [58,59]. Although the lack of shared haplotypes may be attributed to the small sample size in some analysis groups [60], the distinct genetic groups—between H. bispinosa and R. microplus, as well as between R. microplus clades A and C—remain supported by the results of phylogenetic inference and network analysis. The haplotype diversity and nucleotide diversity of H. bispinosa and R. microplus clades A and C, based on the samples collected in this study, represent different categories of genetic diversity. H. bispinosa exhibited low values for both haplotype diversity (Hd < 0.5) and nucleotide diversity (Pi < 0.5), suggesting this tick species recently experienced a population bottleneck or a founder effect [61]. Conversely, R. microplus clades A and C showed high values of haplotype diversity (Hd > 0.5) and low values of nucleotide diversity (Pi < 0.5), presumably due to recent expansion of this tick population [62].
Although population expansion of ticks is commonly associated with recent cattle trade flows that facilitate the spread of ticks through live cattle transfers, the skyline plot in the present study suggested an expansion began over 25,000 years ago, predating modern trade routes. The initiation of the expansion period for R. microplus was estimated to have occurred during the late Pleistocene epoch, approximately 129,000–11,700 years ago [63]. During this period, the region that is now Thailand served as a major route for human migration and the dispersal of various mammalian species [64–66]. The high population density and migratory activities of both hominin and non-hominin fauna, including Artiodactyla, were likely significant factors in driving the population growth of R. microplus during this time period.
R. microplus exhibited relatively high haplotype (Hd > 0.5) and low nucleotide (Pi < 0.5) diversities in both regional and overall analyses across Thailand, suggesting the R. microplus population in Thailand is undergoing expansion. A similar pattern of genetic diversity has been observed in tick populations globally, including Malaysia [55], Brazil [50] and the Neotropical region covering Panama, Colombia, Brazil, and Argentina [18]. These observed genetic characteristics indicated these tick populations are expanding globally as well as in Thailand.
The population structure of R. microplus clade A revealed distinct differences between the peninsular (southern) and mainland populations. The lack of genetic structure and high gene flow observed among mainland populations (northern, northeastern, and central regions) in this study is likely influenced by the intensive production and movement of cattle and buffalo across these areas. Additionally, connections to adjacent countries likely contributed indirectly to increased gene flow between tick populations [67]. Limited gene flow between the southern and other regions could be due to less economic activity and thus less movement of cattle and buffalo connected to the southern region [68–70]. Because genetic differences were detected between mainland and peninsular populations, further study of R. microplus indigenous to southern Thailand is warranted.
In summary, this report confirmed the utility of integrating morphological and molecular approaches for identification of tick species indigenous to Thailand. The use of mitochondrial COI sequencing not only rectified challenges of classifying morphologically similar Haemaphysalis spp. but also provided finer resolution at the subspecies level of R. microplus. These findings confirmed that R. microplus is the predominant species affecting cattle in Thailand, with clade A widespread among multiple regions and clade C limited to the north, while H. bispinosa was established in the northeast. Haplotype analyses revealed distinct genetic groups with high haplotype but low nucleotide diversities within R. microplus clades, indicating an expansion tracing back to the late Pleistocene epoch, a period marked by significant faunal migrations. Collectively, these results underscore the economic impact of tick infestations on Thailand’s livestock sector and highlight potential risks associated with live cattle trade, which may influence the distribution of R. microplus clades. Further research, particularly into lineages and regional genetic structures, is warranted to develop effective management strategies for mitigation of tick-borne diseases of livestock in Thailand.
Supporting information
S1 TableReference sequences of R. microplus, Rhipicephalus spp., Haemaphysalis spp. and outgroup (Ixodes ricinus).(DOCX)
S2 TableHaplotype distribution of H. bispinosa and R. microplus Clades A and C in Thailand.(DOCX)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jonsson NN. The productivity effects of cattle tick (Boophilus microplus) infestation on cattle, with particular reference to Bos indicus cattle and their crosses. Vet Parasitol. 2006;137(1–2):1–10. doi: 10.1016/j.vetpar.2006.01.010 16472920 · doi ↗ · pubmed ↗
- 2Rodrigues DS, Leite RC. Economic impact of Rhipicephalus (Boophilus) microplus: estimate of decreased milk production on a dairy farm. Arq Bras Med Vet Zootec. 2013;65(5):1570–2.
- 3Steelman CD. Effects of external and internal arthropod parasites on domestic livestock production. Annu Rev Entomol. 1976;21:155–78. doi: 10.1146/annurev.en.21.010176.001103 2093 · doi ↗ · pubmed ↗
- 4Uilenberg G. International collaborative research: significance of tick-borne hemoparasitic diseases to world animal health. Vet Parasitol. 1995;57(1–3):19–41. doi: 10.1016/0304-4017(94)03107-8 7597784 · doi ↗ · pubmed ↗
- 5Ghosh S, Bansal GC, Gupta SC, Ray D, Khan MQ, Irshad H, et al. Status of tick distribution in Bangladesh, India and Pakistan. Parasitol Res. 2007;101 Suppl 2:S 207-16. doi: 10.1007/s 00436-007-0684-7 17823830 · doi ↗ · pubmed ↗
- 6Roy BC, Estrada-Peña A, Krücken J, Rehman A, Nijhof AM. Morphological and phylogenetic analyses of Rhipicephalus microplus ticks from Bangladesh, Pakistan and Myanmar. Ticks Tick Borne Dis. 2018;9(5):1069–79. doi: 10.1016/j.ttbdis.2018.03.035 29661691 · doi ↗ · pubmed ↗
- 7Tanskul P, Stark HE, Inlao I. A checklist of ticks of Thailand (Acari: Metastigmata: Ixodoidea). J Med Entomol. 1983;20(3):330–41. doi: 10.1093/jmedent/20.3.330 6876093 · doi ↗ · pubmed ↗
- 8Sarataphan N, Kakuda T, Chansiri K, Onuma M. Survey of benign Theileria parasites of cattle and buffaloes in Thailand using allele-specific polymerase chain reaction of major piroplasm surface protein gene. J Vet Med Sci. 2003;65(1):133–5. doi: 10.1292/jvms.65.133 12576720 · doi ↗ · pubmed ↗