First report of neonatal-onset glutaric aciduria type II in the Iranian population caused by a novel deleterious ETFA variant
Farshid Parvini, Mobarakeh Ajam-Hosseini, Marziyeh Shadpour

TL;DR
This paper reports the first case of a rare metabolic disorder called glutaric aciduria type II in Iran, caused by a new genetic variant in the ETFA gene.
Contribution
The study identifies a novel ETFA gene variant associated with neonatal-onset glutaric aciduria type II in the Iranian population.
Findings
A novel heterozygous in-frame variant c.485_493del in the ETFA gene was identified in an Iranian family.
The ETFA variant co-segregated with the autosomal recessive GA2 disorder in the family.
Bioinformatics analysis confirmed the pathogenicity of the ETFA variant.
Abstract
Glutaric acidemia type II (GA2), also known as multiple acyl-CoA dehydrogenase deficiency (MADD), is a rare inherited error of amino acid and fatty acid metabolism. Its clinical manifestations can vary from severe events that threaten the life of a newborn to milder and late manifestations. Here, we examined an Iranian couple for pre-pregnancy counseling who had a history of the death of two children suspected of metabolic disorder. Whole exome sequencing (WES) was performed to determine possible pathogenic genes in the parents of two deceased neonates. Sanger sequencing was then used to confirm the variant found. Subsequently, the possible impact of the identified variant on the ETFA protein was evaluated using bioinformatics tools. WES identified a novel heterozygous in-frame variant c.485_493del: p.E162_T164del in exon 6 of the ETFA gene, which co-segregated with the autosomal…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
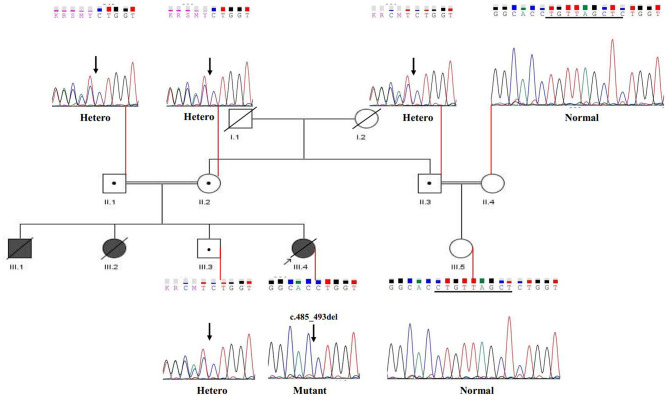 Figure 1
Figure 1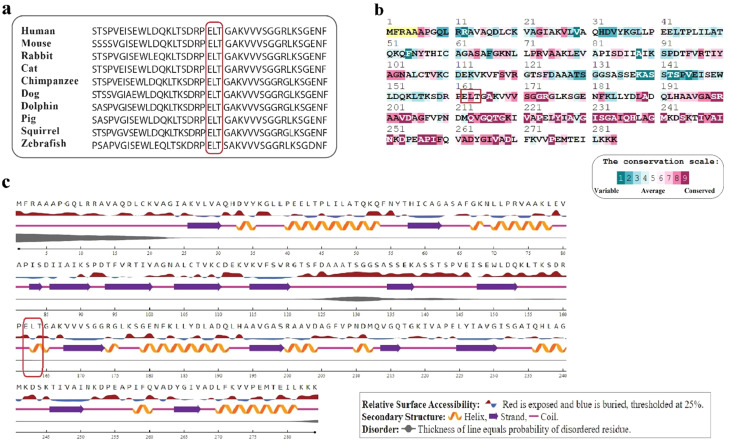 Figure 2
Figure 2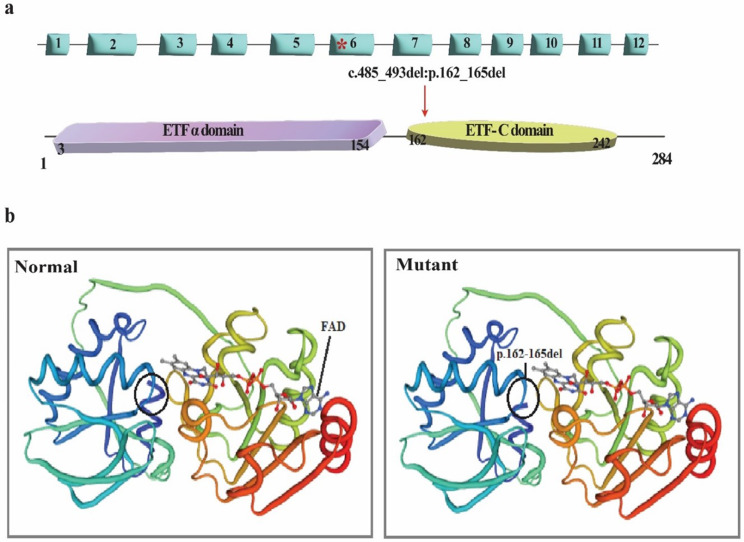 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMetabolism and Genetic Disorders · Mitochondrial Function and Pathology · RNA and protein synthesis mechanisms
Background
Glutaric acidemia type II (GA2) or acyl-CoA dehydrogenase deficiency (MADD) is a rare congenital metabolic disorder with autosomal recessive inheritance caused by a defect in mitochondrial beta-oxidation and inefficient metabolism of amino acids and choline [1]. The GA2 frequency is estimated to be about 1/200,000 live births [2]. Electron transport flavoprotein (ETF) is a dimeric mitochondrial matrix protein composed of two subunits, alpha (ETFA) and beta (ETFB). ETFA protein has two domains and a FAD cofactor, which is essential for maintaining mitochondrial flavin balance [3]. GA2 is a heterogeneous disease. Several genes have been implicated in GA2 disease, including ETFA, ETFB, and ETF dehydrogenase (ETFDH) [4], which were mapped to 15p23–25, 19q13.1 and 4q33, respectively [5]. In 1977, Ruzicka et al., first demonstrated that ETF-QO acts as an electron acceptor for ETF and ubiquinone reductase [6]. ETF is an electron acceptor for dehydrogenases, and along with ETF-QO provide a short path to transfer electrons from several different mitochondrial adenine dinucleotides containing mitochondrial acyl-CoA dehydrogenases to ubiquinone [7]. Ubiquinone is located in the inner membrane of mitochondria and plays a role in ATP production in the respiratory chain [8].
In the electron transfer pathway, 18 enzyme defects have been reported, and the related disorders and transport proteins are known as fatty acid oxidation disorders. These disorders, with the mechanism of autosomal recessive inheritance, lead to a wide range of symptoms related to the lack of energy in the tissues related to the oxidation of fatty acids, which have sometimes been observed in periods of metabolic crisis [9, 10]. So far, numerous mutations in the ETFA, ETFB, and ETFDH genes have been reported with variable clinical manifestations, which can be classified into three categories: (I) neonatal onset with congenital abnormalities, (II) neonatal onset without congenital anomalies, and (III) late-onset with myopathic phenotype, vomiting, hypoglycemia and rarely metabolic acidosis [11, 12]. Although the genotype-phenotype correlation is significant in GA2 types I and II, it appears poor in type 3. Type I is usually linked to homozygous null mutations, whereas type II is often due to mutations that cause amino acid substitutions, enabling a certain level of remaining enzyme activity [3].
Most neonatal-onset GA2 patients die due to severe postnatal abnormalities such as respiratory failure, cardiomyopathy, hypotension, metabolic acidosis, and profound hypoglycemia [13]. Patients with milder or later-onset disease had more successful treatment than patients with neonatal onset [14], as most GA type I and II patients did not survive due to progressive deterioration [15]. In this study, we conducted a genotypic and phenotypic analysis of a patient with GA2, which resulted in identifying a novel pathogenic variant in the ETFA gene. Such studies emphasize the significant role of the whole exome sequencing as a robust technique for diagnosing patients with this heterogenous inherited disorders [16–19].
Materials and methods
Ethical consideration and data collection
Ethical approval for data collection was obtained from the Ethics Committee of the Pharmaceutical Sciences Unit of Islamic Azad University, Tehran, Iran (Ethical Approval Code No. IR.IAU.PS.REC.1396.91) and written informed consent was obtained from the legal guardian.
Patients
Here, we investigated an Iranian family located in Semnan province (central Iran). As shown in Fig. 1, parents (II-1 and II-2) of two deceased neonates (III-1 and III-2) are consanguineous couples who are referred for diagnostic and pre-pregnancy counseling. They had an apparently healthy 17-year-old boy and a history of the death of two neonates, a girl and a boy, 12 and 15 days after birth. Neonates underwent physical and clinical evaluations. Although they appeared physically healthy at birth, they exhibited multiple clinical abnormalities, including liver dysfunction, mild renal abnormalities, elevated urinary organic acid levels, and distinct body odor, and a significant decrease in blood sugar following breast milk consumption. Due to the limitations in gathering data, the biochemical data for the female newborn is detailed in Table 1.
Fig. 1. Family pedigree and segregation analysis of c.485_493del: p.E162_T164del variant in the ETFA gene. The parents (II.1 and II.2), a healthy son (III.3), and a relative (II.3) are heterozygous carriers of the identified variant in the ETFA gene. The fetus (III.4) was reported to be homozygous for this variant. The sequencing illustrates the normal allele in unaffected individuals (II.4 and III.5), as well
Table 1. Biochemical and urinary laboratory tests of 2-day newborn III.2Biochemistry testClinical resultsNormal valuesAST (SGOT)127 ↑Up to 31 U/LALT (SGPT)57 ↑Up to 31 U/LCreatinine1.5 ↑0.3–0.7 mg/dlGamma-GT212 ↑Woman < 32 lu/LCPK1160 ↑New born before 5 days:195–700 U/LAmmonia172 ↑11–51 micromol/lBlood Glucose21 ↓70–105 mg/dlLDH2920 ↑Up to 1100 Iu/LBUN55–20 mg/dlBill.T8.2 ↑0.1–1.2 mg/dlCa^2+^6.8 ↓8.6–10.3 mg/dlNa^+^145135–145 meq/LK^+^2.5 ↓New born: 3.7-5. 9 meq/LAlbumin3.3 ↓3.5–5.2 gr/dlPH7.2 ↓7.4 Urine analysis
Clinical results
(mmol/mol creatinine)
Normal values Glutaric acid24,612 ↑Normal < 19.1 mmol/mol2-Hydroxyglutaric acid500 ↑Normal < 97.3 mmol/mol3-β-Hydroxybutyric acid101 ↑Normal < 9.8 mmol/mol3-Hydroxyisovaleric acid146 ↑Normal < 43.2 mmol/mol4-Hydroxyphenyllactic acid19,000↑Normal < 11.5 mmol/mol2-Hydroxyisovaleric acid240↑Normally not detectedLactic acid56,000 ↑Normal < 564 mmol/molAST; Aspartate aminotransaminase, ALT; Alanine aminotransferase, CPK; Creatine phosphokinase, LDH; lactate dehydrogenase, BUN; Blood urea nitrogen, Bill.T; Bilirubin Total
Based on the unusual biochemical results (especially hypoglycaemia and acidosis), the newborns are suspected of having a metabolic disorder, so we proceeded with additional investigations to resolve the matter. In addition, the patient’s ammonia concentration was elevated (172 µmol/L). Based on the biochemical profile, liver function was abnormal, as shown by increased AST (127 U/L) and ALT (57 U/L), although abdominal ultrasonography revealed a normal liver architecture without evidence of structural dysfunction or acute complications.
Additionally, abdominal ultrasonography demonstrated a normal gallbladder, spleen, and pancreas, with no evidence of focal lesions or biliary dilatation. The portal vein diameter was within normal limits. The left kidney measured 48 mm and the right kidney 51 mm, both slightly enlarged and showing increased cortical echogenicity. Mild fullness was observed in the left renal collecting system, with an anteroposterior diameter of the renal pelvis measuring 3.1–8.2 mm. Mild to moderate hydronephrosis was present on the right side, with a renal pelvic diameter of approximately 3.5 mm. The urinary bladder was empty and therefore not evaluable. No free intraperitoneal fluid was detected.
Brain ultrasound revealed no evidence of intraventricular hemorrhage (IVH), intracranial hemorrhage (ICH), or hydrocephalus. The ventricles appeared mildly prominent but within the expected range for a term neonate, and the sulcal and gyral patterns were normal.
Echocardiographic evaluation demonstrated left ventricular dilatation with an ejection fraction of approximately, indicating reduced systolic function. Valvular abnormalities were identified, including mitral regurgitation (MR) and tricuspid regurgitation (TR). In addition, a patent foramen ovale (PFO) was observed, accompanied by bi-ventricular hypertrophy (BVH) (with left ventricular hypertrophy exceeding right ventricular hypertrophy). The atrial situs was solitus with bi-atrial enlargement, and the ventricles showed a d-loop configuration. The great vessels and coronary anatomy were normal. Importantly, echocardiographic assessment revealed mild pulmonary hypertension (PH), consistent with the hemodynamic alterations.
Fundoscopy revealed normal vascularization with intraocular pressure within the normal range. In the right eye, an area of hyperfluorescent changes was observed near the albinoid fovea. Both eyes demonstrated features consistent with retinopathy of prematurity (ROP), Stage II, Zone III.
According to the clinical symptoms of the neonates, their healthy parents were subjected to genetic testing, for which purpose Whole Exome Sequencing (WES) was performed.
Whole exome sequencing
The karyotype of the parents of two deceased neonates was examined and no chromosomal abnormalities were observed. Then, genomic DNA was extracted from blood samples of seemingly healthy couples following the guidelines of the QIAamp DNA Blood Mini kit (Germany). To explore the genetic etiology, WES was conducted using Illumina Hiseq2000 sequencing technology along with the Agilent SureSelect Human All Exon V7 Kit (Agilent, Santa Clara, USA), targeting all exons of protein-coding genes and several other essential genomic regions. WES was performed for approximately 100 million reads with > 99% sensitivity and > 95% coverage of target regions. After applying various filters, including frequency, intronic regions, 3’-UTR, 5’-UTR, intergenic regions, non-coding segments, and synonymous mutations, the variants were detected.
Computational analysis of variants and Sanger sequencing
Following the approach taken in the previous study [20], a range of bioinformatics tools, including the BWA aligner, Annova, and GATK, were utilized for enhanced accuracy in reading, functional annotation, and increased throughput of the WES results. In addition, public databases ClinVar, gnomAD, Kaviar and GME were also used. The local population database (BayanGene) was utilized to screen 4,000 unrelated healthy individuals (controls), as well. The potential pathogenic impact of the identified variant was assessed using Mutation Taster and Varsome. Additionally, the Phyre2 tool was employed to examine the influence of the variant on the structural alterations of the ETFA protein in comparison to the wild type protein. ClustalW and NetSurfP-2.0 tools were used to align the ETFA protein sequence among 6 different species and check the accessibility of the residues to the surface and secondary structure of the protein. Given the studied parents’ referral for pre-pregnancy counseling and the background of the death of two children with abnormalities (III.1 and III.2), prenatal diagnosis was advised for the fetus (III.4). To validate the novel variant found, the parents, an apparently healthy child, and a fetus were also analyzed using Sanger sequencing, and the results were examined with the Chromas software. Primers F-5’ AACCATACTCTGCAGAATTC 3’ and R-5’ GATTTCTGGAGTCATACAGC 3’ (PCR product: 621 bp) were designed using the Primer3 program. Additionally, to exclude the chromosomal abnormalities, the chorionic villus sample of fetus was scrutinized to examine the STR markers on chromosomes X, Y, 13, 18, and 21 through the Quantitative Fluorescence-Polymerase Chain Reaction (QF-PCR) approach, and the “3130 Genetic Analyzer” and fragment analysis software were applied.
Results
WES results revealed a novel heterozygous in-frame deletion variant in exon 6 of the ETFA gene (NM_0011127716); chr15:76285661: c.485_493del: p.162_165del in the studied parents (II.1 and II.2 in the Fig. 1). The results obtained from the public databases and the local population database (Bayangen) also did not show any previous reports similar to the variant identified in the ETFA gene, which reinforces the novelty of this variant. According to in silico analysis, the novel variant c.485_493del was predicted to be disease causing and likely pathogenesis (Table 2).
Table 2. Prediction of pathogenicity of variant identified in ETFA geneGeneMutationMutationTasterVarsomeACMG Criteria Applied ETFA Pathogenic(NM_0011127716)c.485_493del(E162_T164del)DiseasecausingLikelyPM2, PM4, PP3, PP5
As shown in Fig. 1, Sanger sequencing confirmed the presence of the variant in heterozygous form in the ETFA gene of the parents studied. The healthy child (III.3) and one relative (II.3) were heterozygous carriers for this variant. These findings are consistent with an autosomal recessive inheritance pattern for the disease. This variant leads to the deletion of 9 nucleotides in the coding region of the gene and, given the importance of this region in protein function, is thought to result in a non-functional protein. In order to further investigate this hypothesis, the amino acid residues present in the deletion region (Fig. 2) and the three-dimensional structure of the protein (Fig. 3) were examined.
Fig. 2. Bioinformatics results of the target sequence in ETFA protein. (a) The position of the deleted residues ELT is indicated by a red box. Sequence alignments of ETFA protein in 10 different species indicate high conservation among different organisms. (b) According to ConSurf results, glutamic acid, leucine and threonine residues are in conservation scale 6, 8 and 5, respectively. (c) The secondary structure of the ETFA protein and the location of glutamine, leucine, and threonine amino acids have been predicted by NetSurfP-2.0
Fig. 3. Schematic structure of the ETFA gene/protein and bioinformatic analysis of normal and mutant ETFA protein. (a) The position of the studied variant (c.485_493del: p.E162_T164del) is shown at the gene level (top) with an asterisk and in the protein (bottom). (b) The effect of deletion variant on ETFA protein structure was predicted by Phyre2 software. The variant resulted in a structural change of the ETF-C domain
As shown in Fig. 2a and b, the position of the ELT residues in the ETFA protein is highly conserved, which most likely indicates the pathogenicity of the variant found. Regarding the positioning of residues, the ET and L are found on the surface and depth of the ETFA protein, respectively. In the secondary structure of the protein, the E residue is in the coil and the LT residues are in the alpha helix (Fig. 2c). Therefore, deletion of this sequence is predicted to lead to a significant change in protein structure and function, as shown in Fig. 2.
The identified variant leads to the deletion of three codons encoding amino acids glutamic acid, leucine, and threonine in the C-terminal domain of the ETFA protein (Fig. 3a). To investigate the effect of the c.485_493del mutation on protein structure, we evaluated the three-dimensional structure of the normal and mutant proteins (Fig. 3b). Modeling the structure of the mutant protein revealed that the deletion of amino acids in the 162–165 region (p.162_165del) leads to changes in the ETF-C functional domain of the protein.
Phenotypes observed in deceased children (according to medical records) correspond to the novel identified variant in ETFA gene, and genotype-phenotype correlation studies also confirm it. QF-PCR results showed balanced sets of sex chromosomes and chromosomes 13, 18, and 21 in fetus (III-4). Therefore, fetus is an euploid for the tested chromosomes. The Sanger sequencing confirmed the presence of the variant as heterozygous in the parents (II-1 and II-2), their healthy son (III-3), and a relative (II.3), and as homozygous in the affected fetus (III-4) (Fig. 1). Following the confirmation of the novel variant in the fetus, a legal abortion permit was issued, and the fetus was terminated at the 16th week of its mother’s gestation, with the parents’ consent.
Discussion
In this study, we identified a novel heterozygous non-frameshift deletion (NM_0011127716: exon 6: c.485_493del: p.E162_T164del) in the ETFA gene of the parents of two deceased neonates. Based on the review of the conducted studies, this is the first genetic study of the ETFA gene causing the rare disease GA2 in the Iranian population. Our findings showed that this variant leads to the deletion of three codons and consequently the deletion of three amino acids (ELT; glutamic acid, leucine, and threonine) in the C-terminal region of the ETFA protein (Fig. 3b). Since the deleted amino acids (ELT) are located at conserved positions in the ETFA protein (Fig. 2a and b), and the two amino acids L and T are located in the alpha-helical structure of the protein (Fig. 2c), the deletion of this conserved sequence is expected to lead to a serious disruption of the protein structure and function.
These findings are consistent with previous studies, where researchers have shown through complete multiple alignment of ETF family proteins that the C-terminal region exhibits a high degree of sequence identity compared to the N-terminus indicating the critical significance of the C-terminal region in the functionality of ETF family proteins [21]. Moreover, FAD is situated in the space between the two subunits of the ETF protein, with the majority found within the C-terminal region of the α subunit [22]. Consequently, mutations in the C-terminal region can also affect FAD placement and lead to significant impairment of protein function.
GA2 is a genetic disorder with heterogeneous clinical manifestations, ranging from severe neonatal to mild late-onset forms [23, 24]. Clinical manifestations and disorder onset may vary depending on the location and nature of the mutations [25]. According to studies, Neonatal-onset GA2 is usually fatal and presents with severe metabolic decompensations including non-ketotic hypoglycemia, metabolic acidosis, hypotonia, hyperammonemia, coma, and cardiomyopathy. While the duration and age of late-onset forms are highly variable, in adolescents and adults, muscle or cardiac symptoms or episodic vomiting are generally the first manifestations indicative of GA2 [26, 27]. The measurement and analysis of organic acid in the urine show the accumulation of acid in the blood, which is excreted by the kidneys, and in GA2 patients, increased excretion of lactic and glutaric acids along with other hydroxy and dicarboxylic acids has been reported [28]. GA2 treatment mainly consists of a low-protein, low-fat, high-carbohydrate diet, and supplementation with carnitine and riboflavin to stabilize the complex, which has been associated with greater success [15]. To date, 36 distinct pathogenic and likely pathogenic mutations in the ETFA gene, associated with a variety of abnormalities, have been reported across different regions (Table 3) [3]. In this study, we present the first identification of a novel mutation in Iran, which can serve as a basis for identifying similar patients in different Iranian populations.
Table 3. Clinical characteristics and genotype of patients with pathogenic and likely pathogenic mutations of ETFA geneOriginZygosity/ GenderClinical phenotypeAge at onsetNucleotide sequence(aa changes)Exon/ IntronJapanHetero/ MDyspnea, hypoglycemia, hyperammonemia, respiratory distress, metabolic acidosis, vomiting, fatigue without apparent myopathy0dc.7 C > T(p.R3X)E1GermanyHeteroSevere hypoglycaemia, Cardiac deteriorationDeath: 10 mc.12_22dupE1PortugalHeteroEnergy depletion and impairment of glucose homeostasis, ketogenesisNAc.15_25dup11(p.Q9Rfs20)E1BelgiumHomoNeurological deterioration, Metabolic acidosis, HypoglycemiaDeath: D5c.52 C > T(p.R18X)E2TurkeyHeteroDrowsiness, severe hypoglycaemia, hyperammonemia, metabolic acidosis6 mc.73delA(p.I25X)E2TurkeyHetero/ FErrors of metabolism in asymptomatic newborns, Cystic or dysplastic kidneysnewbornc.200T > C(p.L67P)E3PortugalHeteroBiochemical phenotypeNAc.242 A > C(p.H81P)E3JapanHomo/ MReye-like illness, Poor feeding, CPK elevation, Cardiomyopathy8 mc.283T > G(p.L95V)E4PortugalHeteroBiochemical phenotypeNAc.284T > G(p.L95V)E4PortugalHeteroBiochemical phenotypeNAc.346G > A(p.G116R)E4PortugalNABiochemical phenotypeNAc.347G > T(p.G116V)E4Limoges, FranceHetero/ MFacial dysmorphism, Renalcysts associated with severe hypoglycemia, acidosis, hypotonia, hepatomegaly5yc.354 C > A (p.N118K)E5FranceHomoNA14yc.355dupC(p.L119Pfs10)E5PolynesiaHomoAcute rhabdomyolysis, metabolic acidosis, seizures3yc.365G > A(p.R122K)E5BelgiumHomoNeurological deterioration, metabolic acidosis, hypoglycemiaDeath:D5c.431T > C (p.F144S)E5JapanHomo/ FLiver dysfunction and cardiomyopathy0dIVS6-1G > CI6PortugalHeteroBiochemical phenotypeNAc.453_470del(p.N152_V157del)E6AustriaHomoSevere hypoglycemia, cardiac arrestDeath: D4c.470T > G (p.V157G)E6JapanMChronic fatigue, recurrent vomiting, metabolic acidosis, weakness1yc.478delG(p.D160Mfs*4)E6Iranian(This study)Homo/ FSevere hypoglycemia, hyperammonemia, metabolic acidosis, hypotonia, drowsiness, hepatic dysfunction, cardiac structural and functional abnormalities, mild renal abnormalities, mild brain abnormalities, ophthalmologic abnormalities12dc.485_493del(p.E162_T164del)E6FranceHeteroMetabolic coma with hypoglycemia3 yc.494T > C(p.V165A)E6PortugalHeteroBiochemical phenotypeNAc.502G > T(p.V168F)E6ChineseHetero/ MBiochemical phenotype6 yPortugalHeteroBiochemical phenotypeNAc.563-1G > CE7FranceHomoNA14yc.635T > C(p.L212P)E7Limoges, FranceHetero/ MSevere hypoketotic, hypoglycaemia, hyperammonemia, fatigability, hepatomegaly, energy deficiency5yc.652G > A (p.V218M)E7PortugalHeteroBiochemical phenotypeNAc.664 + 1delE7PortugalHeteroBiochemical phenotypeNAc.667 C > T(p.R223X)E8SpainHomoHepatomegaly, bilateral polycystic kidneys, hypotonia, hypertrophic myocardiopathy, seizures metabolic acidosis, hypoglycemiaDeath: D2c.745 C > T (p.R249C)E9PortugalHeteroBiochemical phenotypeNAc.751G > A(p.A251T)E9JapanHetero/ MReye-like illness, CPK elevation, cardiomyopathy1y764G > T (p.G255V)E9PortugalHeteroBiochemical phenotypeNAc.785 A > G(p.Q262R)E9FranceHeteroSevere hypoglycemia, cardiac ischemia18 mc.797 C > T (p.T266M)E9SenegalHeteroSevere hypoglycemia1yGermanyHeteroSevere hypoglycaemia, cardiac ischemiaDeath:10 mTurkeyHeteroDrowsiness, severe hypoglycaemia, hyperammonemia6 mNetherlandsHomoComa, hypoketotic hypoglycaemia3yTurkeyHetero/ FMyopathic phenotype, chronic fatigue or recurrent vomiting1dJapanHetero/ MDyspnea, hypoglycemia, hyperammonemia0d799G > A (p.G267R)E9PortugalHeteroBiochemical phenotypeNAc.809_811del (p.V270del)E9TurkeyHetero/ FNAnewbornc.854 A > T(p.Q285L)E10FranceHeteroComa, hypoglycemia, myopathic phenotype, rarely metabolic acidosis3yc.875_878delE10PortugalHomoPsychomotor delay, hypotonia, dystonia1yc.963 + 1delGE11
According to the research and Table 3, the majority of pathogenic variants have been identified in exons 9 and 6 of the ETFA gene, suggesting that these exons are prone to mutations [3]. In most cases, ETFA/ETFB/ETF-QO mutations influence the stability of the corresponding messenger RNAs (mRNAs) or the protein network involved in the occurrence of GA2 [29].
Interestingly, hypoglycemic, hyperammonemia, and metabolic acidosis phenotypes have been reported in most patients, and most of them are associated with the neonatal form of GA2. Similarly, in our study, neonates (III.1 and III.2) also showed hypoglycemia, hyperammonemia, metabolic acidosis, drowsiness and hypotonia with strange-smelling secretions without seizures. These findings are consistent with previous reports of patients with neonatal-onset GA2. As previously noted, the majority of GA2 patients with neonatal onset die due to severe abnormalities after birth, in early infancy before GA2 is diagnosed [12]. Considering the irreversible damage of encephalopathic crises and the poor prognosis of acute metabolic crisis in GA1 and GA2 patients, newborn screening is highly recommended for early and correct diagnosis. Current therapeutic mechanisms for GA are based on nutritional therapy [30]. Most GA2 patients have responded well to dietary therapy with L-carnitine, riboflavin or CoQ10.16 supplements [26].
Conclusion
In summary, we identified for the first time a novel deleterious in-frame variant c.485_493del: p.E162_T164del in the ETFA gene of Iranian family. Our study increased the range of ETFA gene variants, provided a better understanding of ETFA variants on phenotypic outcome and reinforced the clinical importance of this gene in patients with neonatal-onset metabolic disorders. As reported in 2003, there is a distinct connection between the phenotype and genotype in GA2 [5]. Therefore, gathering information on genetic mutations to determine any genotype/phenotype correlations and identifying defective enzymes is necessary for accurate prenatal diagnosis of GA2. Further investigations, particularly assessing the potential functional impact of the novel identified mutation could provide deeper insights into the molecular pathogenesis of this mutation.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Mereis M, Wanders RJA, Schoonen M, Dercksen M, Smuts I, van der Westhuizen FH. Disorders of flavin adenine dinucleotide metabolism: MADD and related deficiencies. Int J Biochem Cell Biol. Elsevier Ltd. 2021.10.1016/j.biocel.2020.10589933279678 · doi ↗ · pubmed ↗
- 2Rose L, Hall K, Tang S, Hasadsri L, Kimonis V. Homozygous B 4GALNT 1 mutation and biochemical glutaric acidemia type II: A case report. Clin Neurol Neurosurg. 2020;189.10.1016/j.clineuro.2019.10555331812852 · doi ↗ · pubmed ↗
- 3Chautard R, Laroche-Raynaud C, Lia AS, Chazelas P, Derouault P, Sturtz F, et al. A case report of a mild form of multiple acyl-Co A dehydrogenase deficiency due to compound heterozygous mutations in the ETFA gene. BMC Med Genomics. 2020;13.10.1186/s 12920-020-0665-6PMC 699049031996215 · doi ↗ · pubmed ↗
- 4Henriques BJ, Katrine Jentoft Olsen R, Gomes CM, Bross P. Electron transfer Flavoprotein and its role in mitochondrial energy metabolism in health and disease. Gene. 2021;776.10.1016/j.gene.2021.145407 PMC 794970433450351 · doi ↗ · pubmed ↗
- 5Chokchaiwong S, Kuo YT, Hsu SP, Hsu YC, Lin SH, Zhong W, Bin, et al. ETF-QO mutants uncoupled fatty acid β-oxidation and mitochondrial bioenergetics leading to lipid pathology. Cells. 2019;8.10.3390/cells 8020106 PMC 640655930709034 · doi ↗ · pubmed ↗
- 6Li L, Huang Z, Huang Y, Li Y, Ma X, Li P, et al. Pomalidomide sensitizes lung cancer cells to TRAIL/CDDP-induced apoptosis via directly targeting electron transfer Flavoprotein alpha subunit. Bioorg Chem. 2024;153.10.1016/j.bioorg.2024.10781539265523 · doi ↗ · pubmed ↗
- 7Ou M, Zhu L, Zhang Y, Zhang Y, Zhou J, Zhang Y, et al. A novel electron transfer flavoprotein dehydrogenase (ETFDH) gene mutation identified in a newborn with glutaric acidemia type II: a case report of a Chinese family. BMC Med Genet. 2020;21.10.1186/s 12881-020-00995-2PMC 721258832393189 · doi ↗ · pubmed ↗
- 8Rayat S, Morovvati S. A novel mutation in the glutaryl-Co A dehydrogenase gene (GCDH) in an Iranian patient affected with glutaric acidemia type 1. Clin Case Rep. 2021;9.10.1002/ccr 3.4749 PMC 842308334512980 · doi ↗ · pubmed ↗