Exploring diarylheptanoid derivatives to target LIMK1 as potential agents against colorectal cancer
Liang-Chieh Chen, Tung-Cheng Chang, Hui-Ju Tseng, Jung-Chun Chu, Yun-Yi Huang, Hao-Yuan Peng, Yi-Chen Kuo, Yi-Wen Wu, Tony Eight Lin, Shih-Chung Yen, Kai-Cheng Hsu, Wei-Jan Huang, Shiow-Lin Pan

TL;DR
Researchers designed diarylheptanoid compounds that inhibit LIMK1, a protein linked to colorectal cancer, and found one compound that shows strong potential for treating the disease.
Contribution
A new diarylheptanoid scaffold is proposed as a promising lead for LIMK1 inhibition in colorectal cancer treatment.
Findings
Compound 13a inhibited LIMK1 with an IC50 of 0.94 µM and showed selectivity for tyrosine kinase-like family members.
Compound 13a induced cell cycle changes in CRC cells, suggesting apoptosis induction.
Catechol-containing diarylheptanoids are identified as a promising scaffold for LIMK1 inhibitors.
Abstract
LIMK1 has been demonstrated to be highly correlated with the progression and overall survival rates of colorectal cancer (CRC) patients. In this study, a series of diarylheptanoid scaffold derivatives were intentionally designed and synthesised to evaluate their potential as LIMK1 inhibitors. Among these compounds, compounds 13a and XV exhibited LIMK1 inhibitory activity with IC50 values of 0.94 and 0.57 µM, respectively. We also disclosed the structure–activity relationship of the resulting compounds that exhibited LIMK1 inhibition. Catechol-containing diarylheptanoid was identified as a promising scaffold for LIMK1 inhibitors. Notably, compound 13a demonstrated selectivity in inhibiting the tyrosine kinase-like family and exhibited potent inhibition of CRC cells. Moreover, compound 13a induced an increase in the S phase and a decrease in the G0/G1 phase in a dose-dependent manner,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
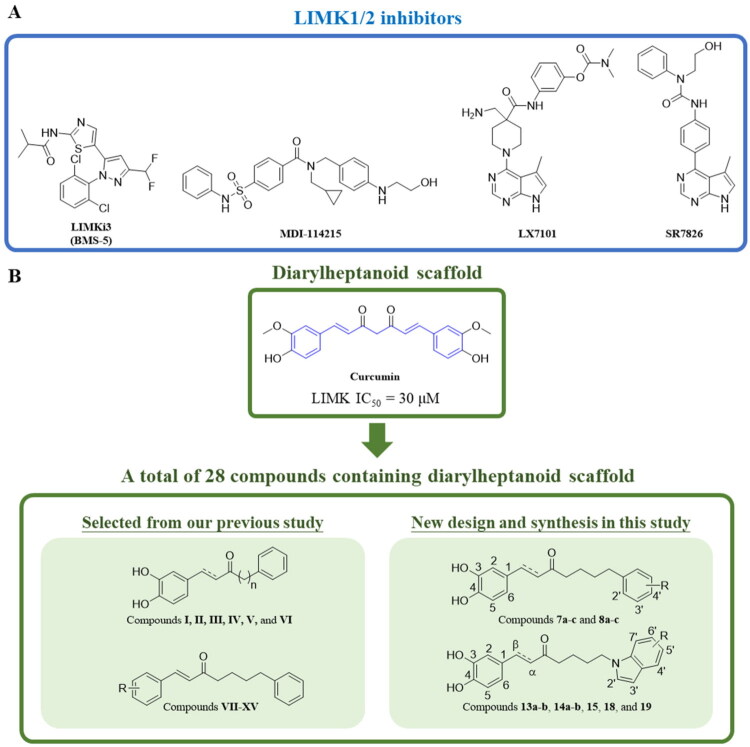 Figure 1
Figure 1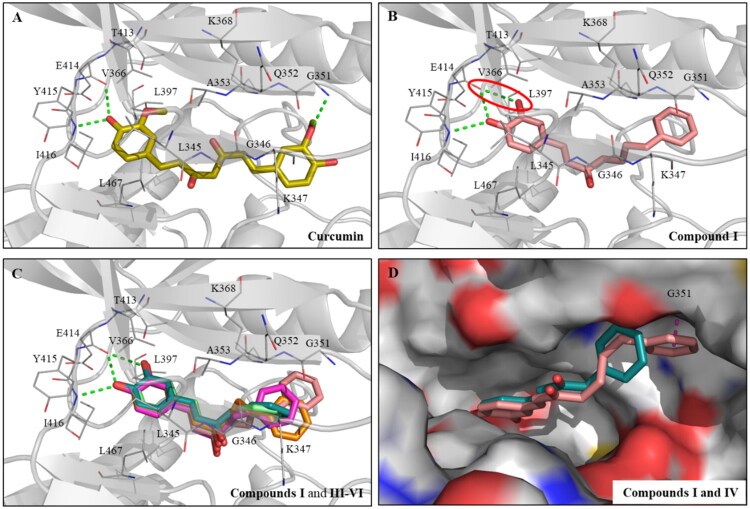 Figure 2
Figure 2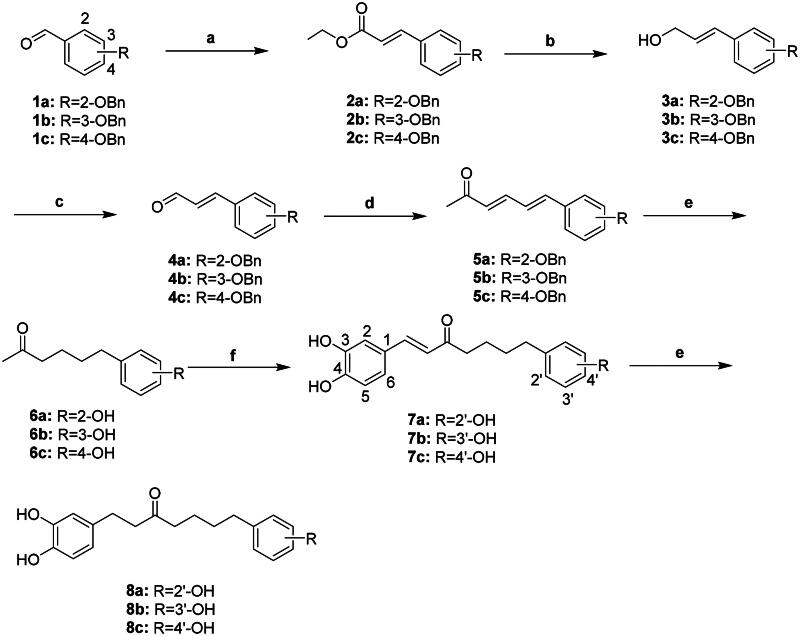 Scheme 1
Scheme 1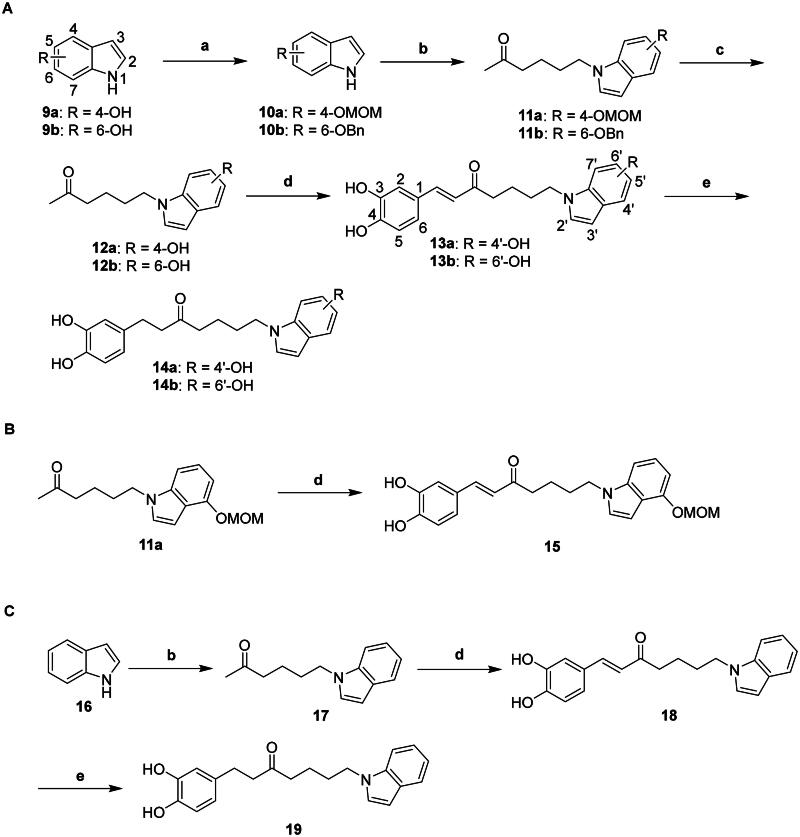 Scheme 2
Scheme 2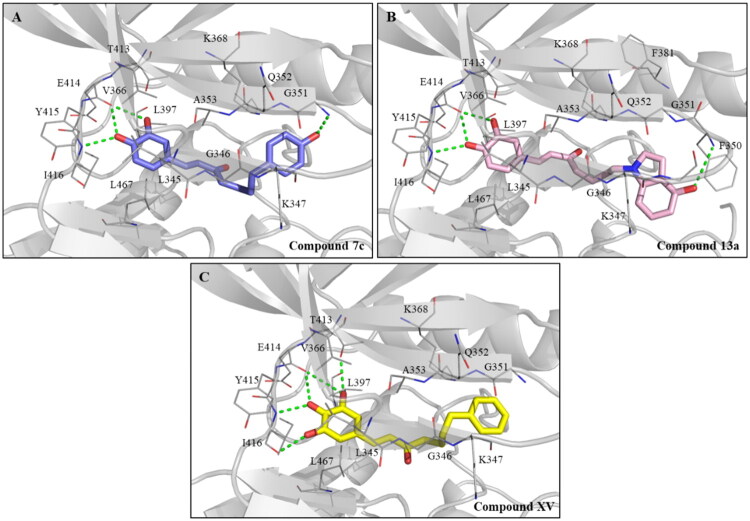 Figure 3
Figure 3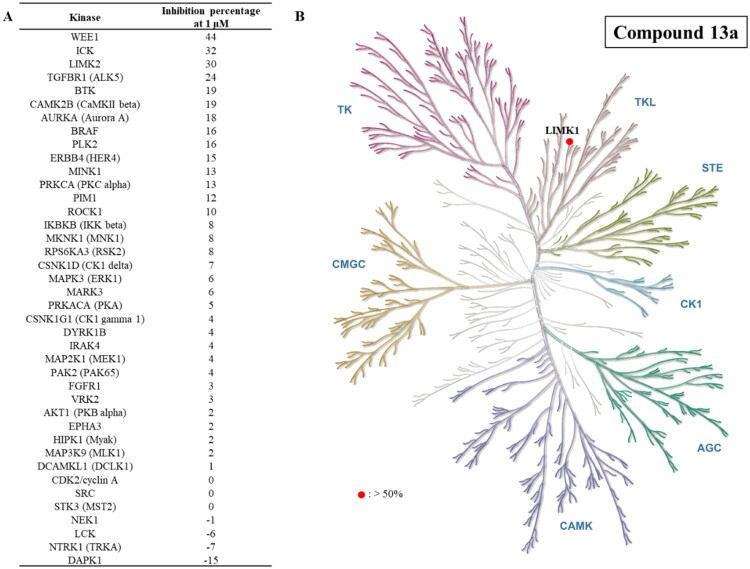 Figure 4
Figure 4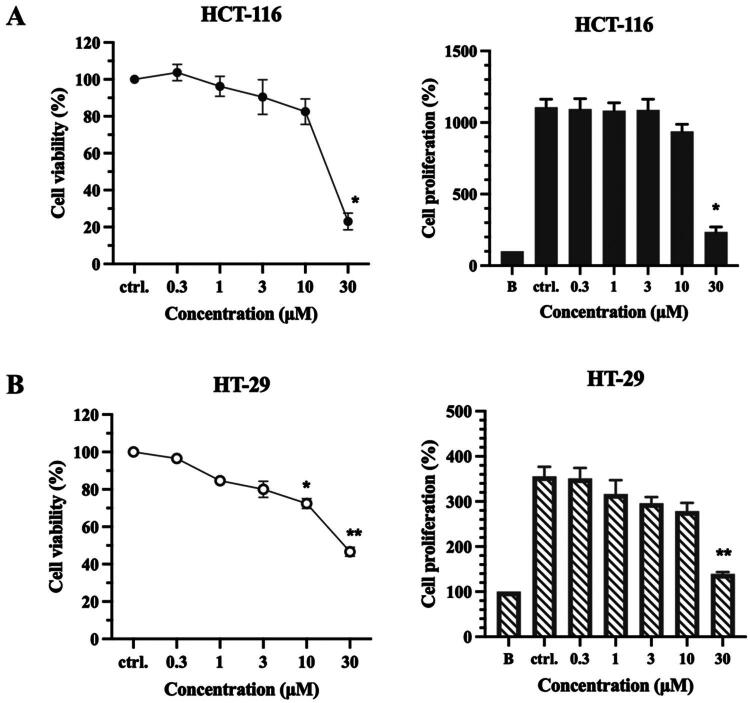 Figure 5
Figure 5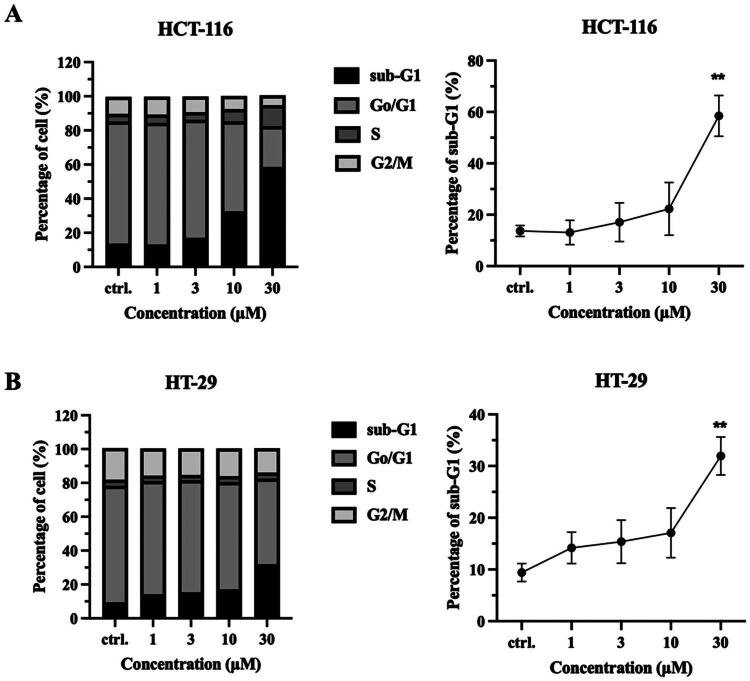 Figure 6
Figure 6|
| |||
|---|---|---|---|
| Compound | Chain length ( | α, β-saturation | Inhibition percentage at 10 μM (IC50) |
|
| 4 | Unsaturated | 101% (1.59 µM) |
|
| 4 | Saturated | 30% |
|
| 0 | Unsaturated | 84% |
|
| 1 | Unsaturated | 56% |
|
| 2 | Unsaturated | 68% |
|
| 3 | Unsaturated | 82% |
|
| ||||
|---|---|---|---|---|
| Compound | Substitution (R) | α, β-saturation | Inhibition percentage at 10 μM | IC50 |
|
| 2′-OH | Unsaturated | 89% | 1.77 µM |
|
| 3′-OH | Unsaturated | 102% | 1.11 µM |
|
| 4′-OH | Unsaturated | 106% | 1.09 µM |
|
| 2′-OH | Saturated | 37% | N.D. |
|
| 3′-OH | Saturated | 34% | N.D. |
|
| 4′-OH | Saturated | 47% | N.D. |
|
| 4′-OH | Unsaturated | 109% | 0.94 µM |
|
| 6′-OH | Unsaturated | 106% | 1.39 µM |
|
| 4′-OH | Saturated | 51% | N.D. |
|
| 6′-OH | Saturated | 53% | N.D. |
|
| 4′-OMOM | Unsaturated | 106% | 1.88 µM |
|
| H | Unsaturated | 110% | 1.49 µM |
|
| H | Saturated | 28% | N.D. |
|
| ||
|---|---|---|
| Compound | Substitution (R) | Inhibition percentage at 10 μM (IC50) |
|
| H | ≤0% |
|
| 4-OH | ≤0% |
|
| 3-OH | ≤0% |
|
| 3-F, 4-OH | ≤0% |
|
| 3-Cl, 4-OH | 4% |
|
| 3-Br, 4-OH | 1% |
|
| 3-NO2, 4-OH | 7% |
|
| 3-OCH3, 4-OH | 1% |
|
| 3,4,5-OH | 112% (0.57 µM) |
| Compounds | HCT-116 | HT-29 | ||
|---|---|---|---|---|
| IC50 (μM) | GI50 (μM) | IC50 (μM) | GI50 (μM) | |
|
| 18.24 ± 0.9 | 14.65 ± 0.8 | 28.78 ± 0.4 | 10.36 ± 0.6 |
|
| > 30 | > 30 | > 30 | 28.77 ± 1.8 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCurcumin's Biomedical Applications · Cell Adhesion Molecules Research · Protein Tyrosine Phosphatases
Introduction
The LIM kinase family consists of two members, LIMK1 and LIMK2, collectively known as LIMK1. Both kinases have two N-terminal LIM domains, a PDZ domain connected to proline/serine-rich regions, and a C-terminal kinase domain1^,^2. The LIM domains are protein-binding motifs that interact with substrates, such as cofilin1, cofilin2, and actin-depolymerising factor (ADF)1^,^3. ADF/cofilin family can bind to actin and regulate actin filament dynamics, which is essential for cell migration, morphogenesis, and cell division4. Active cofilin/ADF can convert filamentous actin (F-actin) into monomeric globular actin (G-actin), promoting actin turnover and modulating cell motility. In contrast, inactivated cofilin/ADF loses its function, leading to the accumulation of actin polymers, stabilising actin filaments, and suppressing actin turnover4^,^5. LIMK can phosphorylate cofilin/ADF on serine 3, leading to cofilin inactivation and inhibition of its function6^,^7. Through regulating cofilin activity, LIMK can modulate actin cytoskeletal dynamics during cell migration8.
LIMK1 is overexpressed and essential in various types of cancer9–12. Studies have indicated that overexpressed LIMK1 can boost the invasion, metastatic ability, and progression of cancer cells12–15. Protein kinase C ζ (PKCζ) knockdown is linked to reduced phosphorylation of LIMK1 and cofilin, leading to the inhibition of migration of glioblastoma cells and macrophages16^,^17. Knockdown or inhibition of LIMK1 inhibits the proliferation of lung cancer, gastric cancer, colorectal cancer (CRC), and acute myeloid leukaemia (AML) cells, as well as the invasion motility of glioblastoma11^,^12^,^18–20. Furthermore, LIMK1 knockdown did not exhibit significant toxicity in mouse models of gastric cancer and glioblastoma, suggesting that LIMK1 could be a potential target without serious health concerns in vivo12^,^19.
The expression of LIMK1 is elevated in all stages of CRC21,22 which is one of the leading causes of cancer-related deaths globally. Analysis of 143 CRC samples revealed markedly elevated cytoplasmic expression of LIMK1 (93.7%), LIMK2 (89.5%), and cofilin (86.7%), while nuclear expression was detected in only 54.5%, compared with adjacent adenomas and non-neoplastic epithelium23. These findings suggest that LIMK1, LIMK2, and cofilin are predominantly activated in the cytoplasm of CRC cells, supporting their role in cytoskeletal remodelling and tumour progression. In clinical cohorts, CRC patients with elevated LIMK1 levels exhibit reduced overall survival and heightened lymph node metastasis22. Approximately 20% of CRC patients experience metastasis, and 40% have recurrence after treatment22^,^24^,^25. Metastatic CRC is incurable for most patients and has a less than 20% 5-year survival rate24, highlighting the unmet medical need for CRC treatment. Unlike the decreased role of LIMK2 in CRC initiation, LIMK1 is overexpressed and drives the invasion and migration of CRC cells9^,^21. Elevated LIMK1 expression is significantly associated with adverse clinicopathological features, including lymphatic invasion and advanced pathological stage21. Numerous studies have demonstrated that LIMK1 is highly correlated with the progression and overall survival rates of CRC patients15^,^20–22. Clinical and transcriptomic analyses revealed that LIMK1 is highly expressed at both mRNA and protein levels in CRC21, with expression correlated to lymphatic invasion, advanced pathological stage, poor prognosis, and immune cell infiltration (CD4^+^ T cells, macrophages, and dendritic cells)21. Together, these findings indicate that LIMK1 is a critical driver of CRC progression and a promising therapeutic target.
Recent advances have clarified the landscape of small-molecule LIMK inhibitors. The thiazole derivative LIMKi3 (BMS-5) (Figure 1(A)), originally developed by Bristol-Myers Squibb, is a highly potent dual LIMK1 and LIMK2 inhibitor (IC_50_ = 6 and 33 nM, respectively) that effectively suppresses cofilin phosphorylation and demonstrates efficacy in cancer26 and Fragile X Syndrome27, but its further development has been hindered by poor kinase selectivity26^,^28^,^29. More recently, MDI-114215 (Figure 1(A)) has emerged as a selective and stable LIMK1/2 inhibitor that blocks both nonphosphorylated and PAK1-phosphorylated forms, suppresses cofilin phosphorylation, and rescues synaptic deficits in Fragile X Syndrome models and patient-derived neurons30. The pyrrolopyrimidines LX7101 (developed by Lexicon Pharmaceuticals) and SR7826 (Figure 1(A)) are potent LIMK inhibitors, with LX7101 advancing to clinical trials for glaucoma and SR7826 demonstrating neuroprotective effects in Fragile X and Alzheimer’s models29–31. Despite these promising activities, both compounds remain unselective LIMK1/2 inhibitors, limiting their value as tool molecules29^,^31. Together, these findings underscore the therapeutic promise of LIMK inhibition while highlighting the critical need for LIMK1/2 inhibitors to serve as reliable tool compounds and potential drug candidates.
Representative chemical structures of (A) reported LIMK1/2 inhibitors and (B) the diarylheptanoid-based compound evaluated in this study.
Curcumin, an extensively researched secondary plant metabolite found in turmeric, exhibits significant anti-tumour effects in CRC or other cell lines, including cytotoxic effects, inhibited colony formation, and decreased cell motility32–36. Inhibition of cell motility by curcumin is mediated by reducing levels of active cofilin32. In a separate study, curcumin shows impressive LIMK inhibition with an IC_50_ value of 30 μM37. The main scaffold of curcumin is a diarylheptanoid that consists of 1,7-diphenylheptane and displays a wide variety of bioactivity38^,^39. The linear structure and multiple modified structural positions of diarylheptanoid provide abundant information for exploring the structure–activity relationship (SAR) study. Furthermore, the above study may connect and unveil the potential of diarylheptanoid scaffold derivatives for CRC and present promising opportunities for further research and applications. However, curcumin adopts a relatively rigid and extended conformation, which may restrict its ability to access critical binding residues. In contrast, the diarylheptanoid scaffold provides greater conformational flexibility, which can facilitate more favourable interactions with the target protein.
In this study, we designed and synthesised a series of diarylheptanoid scaffold derivatives and assessed their ability to inhibit LIMK1 (Figure 1(B)). 28 diarylheptanoid scaffold derivatives were assessed for their ability to inhibit LIMK1. Among them, 15 compounds were obtained from previous studies37. Besides that, we synthesised 13 compounds by adding a hydroxy group, which potentially forms additional interactions with LIMK1. To better understand the binding mechanism between LIMK1 and the compounds, we conducted docking and interaction analyses. Among the synthesised diarylheptanoids, the most potent compound was selected to assess its selectivity for other kinases. Additionally, its biological activity was evaluated through anti-proliferative assays and cell cycle analysis. Overall, these findings highlight that structurally modified diarylheptanoids can enhance LIMK1 inhibitory activity, providing additional options for optimising novel LIMK1 inhibitors.
Materials and methods
General procedures
Nuclear magnetic resonance spectra (^1^H and ^13^C NMR) were obtained using Bruker Fourier 300, Avance DRX 500, and AVIII 500 spectrometers. The chemical shifts were reported in parts per million (ppm, δ) with an internal standard, tetramethylsilane (TMS). The mass spectra were measured on THERMO Q Exactive Plus and Finnigan LCQ mass spectrometry. The melting points were determined on an FARGO MP-2D digital melting point apparatus. The purity of final compounds was determined using HPLC with a C18-column (150 mm 4.6 mm, Ascentis) and an L-2130 pump (Hitachi, Ibaraki, Japan). Column chromatography was performed using silica gel (70–230 mesh, Merck, Darmstadt, Germany). The thin-layer chromatography analysis was performed using silica gel plates (KG60-F254, Merck). Unless otherwise mentioned, all chemicals were used without any purification, and all reactions were carried out under an atmosphere of dry nitrogen.
(E)-Ethyl 2-benzyloxycinnamate (2a)
To a solution of NaH (2.26 g, 94.16 mmol) in dry tetrahydrofuran (THF) (50 ml) was added triethyl phosphonoacetate (18.7 ml, 94.25 mmol) in an ice bath. After stirring under N_2_ for 1 h, the resultant was added a solution of 2-benzyloxybenzaldehyde 1a (10.0 g, 47.12 mmol) in dry THF and stirred for an additional 1 h at RT. The reaction mixture was diluted with distilled H_2_O and neutralised with 1 N HCl_(aq)_ to pH = 7. The mixture was extracted with EtOAc (3 50 ml). The combined organic layer was dried over anhydrous MgSO_4_, filtered, and the solvent was concentrated in vacuo. The residue was purified over a silica gel column eluted with 10% EtOAc in n-hexane to give compound 2a (13.12 g, 99%) as a white solid. Melting point 50–52 °C. ^1^H NMR (Acetone-d6, 300 MHz): δ 8.06 (d, J = 16.1 Hz, 1H), 7.70 (dd, J = 7.7, 1.7 Hz, 1H), 7.52 (m, 2H), 7.38 (m, 4H), 7.18 (dd, J = 8.3, 0.8 Hz, 1H), 7.01 (ddd, J = 8.3, 7.7, 0.8 Hz, 1H), 6.58 (d, J = 16.1 Hz, 1H), 5.26 (s, 2H), 4.19 (q, J = 7.1 Hz, 2H), 1.27 (t, J = 7.1 Hz, 3H). ESI-MS m/z: 283.1 [M + H]^+^.
(E)-ethyl 3-benzyloxycinnamate (2b)
Following the procedure as described for the preparation of compound 2a, the reaction of NaH (2.26 g, 94.16 mmol) in THF (50 ml), triethyl phosphonoacetate (18.7 ml, 94.25 mmol), and 3-benzyloxybenzaldehyde (10.0 g, 47.11 mmol) in THF (50 ml) gave compound 2b (11.00 g, 83%) as a white solid. Melting point 39–42 °C. ^1^H NMR (MeOH-d4, 300 MHz): δ 7.64 (d, J = 16.0 Hz, 1 H), 7.44 (m, 2H), 7.34 (m, 4H), 7.19 (m, 2H), 7.04 (ddd, J = 8.2, 2.5, 1.0 Hz, 1H), 6.49 (d, J = 16.0 Hz, 1H), 5.11 (s, 2H), 4.24 (q, J = 7.1 Hz, 2H), 1.32 (t, J = 7.1 Hz, 3H). ESI-MS m/z: 283.1 [M + H]^+^.
(E)-ethyl 4-benzyloxycinnamate (2c)
Following the procedure as described for the preparation of compound 2a, the reaction of NaH (2.26 g, 94.16 mmol) in THF (50 ml), triethyl phosphonoacetate (18.7 ml, 94.25 mmol), and 4-benzyloxybenzaldehyde (10.0 g, 47.11 mmol) in THF (50 ml) gave compound 2c (13.30 g, 99%) as a white solid. Melting point 68–71 °C. ^1^H NMR (Acetone-d6, 300 MHz): δ 7.63 (m, 3H), 7.49 (m, 2H), 7.37 (m, 3H), 7.08 (ddd, J = 9.3, 2.6 Hz, 2H), 6.39 (d, J = 16.0 Hz, 1H), 5.19 (s, 2H), 4.19 (q, J = 7.1 Hz, 2H), 1.27 (t, J = 7.1 Hz, 3H). ESI-MS m/z: 283.1 [M + H]^+^.
(E)-3-(2-(benzyloxy)phenyl)prop-2-en-1-ol (3a)
To a solution of compound 2a (13.00 g, 46.04 mmol) in dry CH_2_Cl_2_ (100 ml) was added 1.2 M diisobutylaluminium hydride (DIBAL-H) in toluene (76.7 ml, 92.04 mmol) dropwise under −30 °C. The resultant was stirred under N_2_ for 3 h. The reaction mixture was quenched with distilled H_2_O at 0 °C, washed with CH_2_Cl_2_ (5 50 ml), and filtered through a celite pad. The filtrate was dried over anhydrous Na_2_SO_4_, filtered, and the solvent was concentrated in vacuo. The residue was purified over a silica gel column eluted with 20% EtOAc in n-hexane to give compound 3a (10.96 g, 99%) as a white solid. Melting point 49–51 °C. ^1^H NMR (Acetone-d6, 300 MHz): δ 7.51 (m, 3H), 7.40 (m, 2H), 7.33 (m, 1H), 7.20 (ddd, J = 8.2, 7.2, 1.4 Hz, 1H), 7.06 (dd, J = 8.2, 1.0 Hz, 1H), 6.99 (ddd, J = 16.1, 1.7, 1.7 Hz, 1H), 6.92 (ddd, J = 7.4, 7.2, 1.0 Hz, 1H), 6.41 (dt, J = 16.1, 5.4 Hz, 1H), 5.17 (s, 2H), 4.23 (td, J = 5.7, 1.7 Hz, 2H), 3.79 (t, J = 5.7 Hz, 1H). ESI-MS m/z: 223.11 [M − OH]^+^.
(E)-3-(3-benzyloxyphenyl)prop-2-en-1-ol (3b)
Following the procedure as described for the preparation of compound 3a, the reaction of compound 2b (7.40 g, 26.21 mmol) in dry CH_2_Cl_2_ (100 ml) was added 1.2 M DIBAL-H in toluene (43.7 ml, 52.44 mmol) to give compound 3b (6.1 g, 97%) as a light yellow solid. Melting point 56–59 °C. ^1^H NMR (MeOD-d4, 300 MHz): δ 7.44 (m, 2H), 7.37 (m, 2H), 7.30 (m, 1H), 7.21 (dd, J = 7.9, 7.9 Hz, 1H), 7.01 (m, 2H), 6.86 (ddd, J = 8.2, 2.5, 0.7 Hz, 1H), 6.57 (m, 1H), 6.34 (dt, J = 15.9, 5.5 Hz, 1H), 6.34 (s, 2H), 4.21 (dd, J = 5.5, 1.5 Hz, 2H). ESI-MS m/z: 223.11 [M − OH]^+^.
(E)-3-(4-benzyloxyphenyl)prop-2-en-1-ol (3c)
Following the procedure as described for the preparation of compound 3a, the reaction of compound 2c (13.30 g, 47.11 mmol) in dry CH_2_Cl_2_ (100 ml), and 1.2 M DIBAL-H in toluene (78.5 ml, 94.20 mmol) gave compound 3c (10.87 g, 96%) as a white solid. Melting point 112–114 °C. ^1^H NMR (Acetone-d6, 300 MHz): δ 7.48 (m, 2H), 7.39 (m, 3H), 7.33 (m, 2H), 6.97 (d, J = 8.8 Hz, 2H), 6.54 (m, 1H), 6.26 (dt, J = 15.7, 5.4 Hz, 1H), 5.12 (s, 2H), 4.20 (ddd, J = 5.6, 5.5, 1.7 Hz, 2H), 3.75 (t, J = 5.6 Hz, 1H). ESI-MS m/z: 223.11 [M − OH]^+^.
2-Benzyloxycinnamaldehyde (4a)
To a solution of compound 3a (10.86 g, 45.19 mmol) in toluene (100 ml) was added MnO_2_ (39.29 g, 451.92 mmol). The resultant was refluxed and stirred under N_2_ for 2 h. The reaction mixture was filtered through a celite pad, washed with toluene, and the filtrate was concentrated in vacuo. The residue was purified over a silica gel column eluted with 5% EtOAc in n-hexane to give compound 4a (7.50 g, 69%) as a yellow solid. Melting point 63–65 °C. ^1^H NMR (Acetone-d6, 300 MHz): δ 9.68 (d, J = 7.7 Hz, 1 H), 7.96 (d, J = 16.1 Hz, 1 H), 7.74 (dd, J = 7.8, 1.8 Hz, 1H), 7.54 (m, 2H), 7.41 (m, 4H), 7.22 (dd, J = 8.4, 0.8 Hz, 1H), 7.05 (m, 1H), 6.82 (dd, J = 16.1, 7.8 Hz, 1H), 5.29 (s, 2H). ESI-MS m/z: 239.1 [M + H]^+^.
3-Benzyloxycinnamaldehyde (4b)
Following the procedure as described for the preparation of compound 4a, the reaction of compound 3b (9.0 g, 37.45 mmol) in toluene (100 ml) and MnO_2_ (32.56 g, 374.55 mmol) gave compound 4b (7.19 g, 81%) as a yellow solid. Melting point 95–97 °C. ^1^H NMR (MeOH-d4, 300 MHz): δ 9.65 (d, J = 7.8 Hz, 1H), 7.63 (d, J = 15.9 Hz, 1H), 7.45 (m, 2H), 7.32 (m, 6H), 7.10 (ddd, J = 8.1, 2.5, 1.0 Hz, 1H), 6.75 (dd, J = 15.9, 7.8 Hz, 1H), 5.13 (s, 2H). ESI-MS m/z: 239.1 [M + H]^+^.
4-Benzyloxycinnamaldehyde (4c)
Following the procedure as described for the preparation of compound 4a, the reaction of compound 3c (10.86 g, 45.19 mmol) in toluene (100 ml) and MnO_2_ (39.19 g, 450.82 mmol) gave compound 4c (7.32 g, 68%) as a white solid. Melting point 77–79 °C. ^1^H NMR (Acetone-d6, 300 MHz): δ 9.66 (d, J = 7.7 Hz, 1H), 7.70 (m, 2H), 7.62 (d, J = 15.9 Hz, 1 H), 7.50 (m, 2H), 7.38 (m, 3H), 7.12 (m, 2H), 6.66 (dd, J = 15.9, 7.7 Hz, 1H), 5.21 (s, 2H). ESI-MS m/z: 239.1 [M + H]^+^.
General procedure for the synthesis of compounds 5a–c
To a solution of 1-(triphenylphosphoranylidene)-2-propanone (31.0 mmol) in dry toluene (50 ml) was added a solution of compounds 4a–c (31.0 mmol) in dry toluene (50 ml). The resultant was refluxed and stirred under N_2_ for 12 h. The reaction mixture was concentrated in vacuo, extracted with EtOAc (200 ml), and washed with distilled H_2_O (3 100 ml). The organic layer was dried over anhydrous MgSO_4_, filtered, and the solvent was concentrated in vacuo. Compounds 5a–c were obtained without further purification.
6-(2-Hydroxyphenyl)hexan-2-one (6a)
To a solution of compound 5a (2.00 g, 7.19 mmol) in EtOH (15 ml) was added 10% Pd–C (400 mg). The resultant was stirred at room temperature under H_2_ atmosphere for 6 h. The reaction mixture was filtered through a celite pad, and the filtrate was concentrated in vacuo. The residue was purified over a silica gel column eluted with 10% EtOAc in n-hexane to give compound 6a (1.38 g, 99%) as a brown solid. Melting point 35–37 °C. ^1^H NMR (Acetone-d6, 300 MHz): δ 8.10 (s, 1H), 7.08 (dd, J = 7.4, 1.8 Hz, 1H), 6.99 (ddd, J = 7.9, 7.5, 1.8 Hz, 1H), 6.80 (dd, J = 7.9, 1.2 Hz, 1H), 6.74 (ddd, J = 7.5, 7.4, 1.2 Hz, 1H), 2.61 (m, 2H), 2.47 (m, 2H), 2.06 (s, 3H), 1.58 (m, 4H). ESI-MS m/z: 193.1 [M + H]^+^.
6-(3-Hydroxyphenyl)hexan-2-one (6b)
Following the procedure as described for the preparation of compound 6a, the reaction of compound 5b (2.00 g, 7.19 mmol) in EtOH (15 ml), and 10% Pd–C (400 mg) gave compound 6b (1.38 g, 99%) as a brown solid. Melting point 75–78 °C. ^1^H NMR (Acetone-d6, 300 MHz): δ 8.13 (s, 1H), 7.07 (m, 1H), 6.65 (m, 3H), 2.53 (m, 2H), 2.47 (m, 2H), 2.06 (s, 3H), 1.56 (m, 4H). ESI-MS m/z: 193.1 [M + H]^+^.
6-(4-Hydroxyphenyl)hexan-2-one (6c)
Following the procedure as described for the preparation of compound 6a, the reaction of compound 5c (2.0 g, 7.19 mmol) in EtOH (15 ml) and 10% Pd–C (400 mg) gave compound 6c (1.38 g, 99%) as a white solid. Melting point 47–50 °C. ^1^H NMR (dimethyl sulfoxide-d6 (DMSO-d6), 300 MHz): δ 9.08 (s, 1H), 6.95 (d, J = 8.5 Hz, 2H), 6.65 (d, J = 8.5 Hz, 2H), 2.43 (m, 4H), 2.05 (s, 3H), 1.45 (m, 4H). ESI-MS m/z: 193.1 [M + H]^+^.
1-(3.4-Dihydroxyphenyl)-7-(2-hydroxyphenyl)hept-1-en-3-one (7a)
To a solution of compound 6a (400 mg, 2.08 mmol) in THF (20 ml) was added pyrrolidine (171 μl, 2.08 mmol), acetic acid (119 μl, 2.08 mmol), and 3,4-dihydroxybenzaldehyde (287 mg, 2.08 mmol). The resultant was refluxed and stirred under N_2_ for 4 h. The reaction mixture was diluted with EtOAc (100 ml) and washed with distilled H_2_O (3 50 ml). The organic layer was dried over anhydrous Na_2_SO_4_, filtered, and the solvent was concentrated in vacuo. The residue was purified over a silica gel column eluted with 2.5% MeOH in CH_2_Cl_2_ to give compound 7a (182 mg, 28%) as a brown solid. Melting point 158–162 °C. ^1^H NMR (Acetone-d6, 500 MHz): δ 8.48 (br, 1H), 8.19 (br, 2H), 7.47 (d, J = 16.1 Hz, 1H), 7.17 (d, J = 2.2 Hz, 1H), 7.10 (dd, J = 7.4, 1.6 Hz, 1H), 7.05 (dd, J = 8.2, 2.2 Hz, 1H), 6.99 (ddd, J = 7.9, 7.4, 1.6 Hz, 1H), 6.86 (d, J = 8.2 Hz, 1H), 6.81 (dd, J = 7.9, 1.1 Hz, 1H), 6.74 (ddd, J = 7.4, 1.1 Hz, 1H), 6.61 (d, J = 16.1 Hz, 1H), 2.66 (m, 4H), 1.66 (m, 4H). ^13^C NMR (Acetone-d6, 125 MHz): δ 200.1, 156.0, 148.8, 146.4, 143.1, 130.9, 129.6, 128.1, 127.7, 124.6, 122.8, 120.3, 116.5, 115.9, 115.3, 40.9, 30.6, 30.4, 25.1. HR-ESI-MS m/z: [M + H]^+^ calcd for C_19_H_21_O_4_ 313.1434, found 313.1434.
1-(3.4-Dihydroxyphenyl)-7-(3-hydroxyphenyl)hept-1-en-3-one (7b)
Following the procedure as described for the preparation of compound 7a, the reaction of compound 6b (150 mg, 0.78 mmol) in THF (10 ml), pyrrolidine (64 μl, 0.78 mmol), acetic acid (45 μl, 0.78 mmol), and 3,4-dihydroxybenzaldehyde (72 mg, 0.52 mmol) gave compound 7b (46 mg, 28%) as a brown solid. Melting point 145–148 °C. ^1^H NMR (Acetone-d6, 500 MHz): δ 8.28 (br, 3H), 7.48 (d, J = 16.1 Hz, 1H), 7.17 (d, J = 2.1 Hz, 1H), 7.07 (m, 2H), 6.87 (d, J = 8.2 Hz, 1H), 6.64 (m, 4H), 2.68 (t, J = 7.0 Hz, 2H), 2.57 (t, J = 7.0 Hz, 2H), 1.65 (m, 4H). ^13^C NMR (Acetone-d6, 125 MHz): δ 199.9, 158.4, 148.8, 146.4, 144.9, 143.1, 130.1, 128.1, 124.6, 122.8, 120.5, 116.5, 116.2, 115.3, 113.6, 40.8, 36.4, 31.9, 24.9. HR-ESI-MS m/z: [M + H]^+^ calcd for C_19_H_21_O_4_ 313.1434, found 313.1434.
1-(3.4-Dihydroxyphenyl)-7-(4-hydroxyphenyl)hept-1-en-3-one (7c)
Following the procedure as described for the preparation of compound 7a, the reaction of compound 6c (500 mg, 2.60 mmol) in THF (20 ml), pyrrolidine (214 μl, 2.60 mmol), acetic acid (149 μl, 2.60 mmol), and 3,4-dihydroxybenzaldehyde (239 mg, 1.73 mmol) gave compound 7c (159 mg, 29%) as a yellow solid. Melting point 148–151 °C. ^1^H NMR (Acetone-d6, 500 MHz): δ 8.21 (br, 3H), 7.47 (d, J = 16.2 Hz, 1H), 7.1 (d, J = 2.1 Hz, 1H), 7.05 (dd, J = 8.2, 2.1 Hz, 1H), 7.02 (d, J = 8.5 Hz, 2H), 6.87 (d, J = 8.2 Hz, 1H), 6.74 (d, J = 8.5 Hz, 2H), 6.60 (d, J = 16.2 Hz, 1H), 2.66 (t, J = 7.0 Hz, 2H), 2.55 (t, J = 7.2 Hz, 2H), 1.62 (m, 4H). ^13^C NMR (Acetone-d6, 125 MHz): δ 200.0, 156.3, 148.8, 146.4, 143.1, 134.0, 130.1, 128.0, 124.6, 122.8, 116.5, 116.0, 115.3, 40.8, 35.5, 32.3, 24.8. HR-ESI-MS m/z: [M + H]^+^ calcd for C_19_H_21_O_4_ 313.1434, found 313.1433.
1-(3.4-Dihydroxyphenyl)-7-(2-hydroxyphenyl)heptan-3-one (8a)
Following the procedure as described for the preparation of compound 6a, the reaction of compound 7a (182 mg, 0.58 mmol) in EtOH (10 ml) and 10% Pd–C (18 mg) gave compound 8a (80 mg, 44%) as a yellow solid. Melting point 123–126 °C. ^1^H NMR (Acetone-d6, 500 MHz): δ 8.12 (s, 1H), 7.69 (s, 1H), 7.64 (s, 1H), 7.07 (dd, J = 7.5, 1.6 Hz, 1H), 6.99 (ddd, J = 7.9, 7.5, 1.6 Hz, 1H), 6.80 (dd, J = 7.9, 1.1 Hz, 1H), 6.74 (ddd, J = 7.5, 7.4, 1.1 Hz, 1H), 6.70 (d, J = 8.0 Hz, 1H), 6.68 (s, J = 2.1 Hz, 1H), 6.51 (dd, J = 8.0, 2.1 Hz, 1H), 2.68 (m, 4H), 2.60 (m, 2H), 2.45 (m, 2H), 1.57 (m, 4H). ^13^C NMR (Acetone-d6, 125 MHz): δ 210.0, 156.0, 145.9, 144.1, 134.2, 130.9, 129.5, 127.7, 120.4, 116.3, 116.1, 115.8, 45.1, 43.1, 30.7, 30.3, 29.9, 24.4. HR-ESI-MS m/z: [M + H]^+^ calcd for C_19_H_23_O_4_ 315.1591, found 315.1591.
1-(3.4-Dihydroxyphenyl)-7-(3-hydroxyphenyl)heptan-3-one (8b)
Following the procedure as described for the preparation of compound 6a, the reaction of compound 7b (80 mg, 0.26 mmol) in EtOH (5 ml) and 10% Pd–C (8 mg) to give compound 8b (42 mg, 52%) as a brown solid. Melting point 98–101 °C. ^1^H NMR (Acetone-d6, 500 MHz): δ 8.12 (s, 1H), 7.67 (s, 1H), 7.63 (s, 1H), 7.07 (t, J = 7.8, Hz, 1H), 6.70 (d, J = 8.0 Hz, 1H), 6.65 (m, 4H), 6.51 (dd, J = 8.0, 2.1 Hz, 1H), 2.68 (m, 4H), 2.51 (m, 2H), 2.44 (m, 2H), 1.55 (m, 4H). ^13^C NMR (Acetone-d6, 125 MHz): δ 209.9, 158.3, 145.8, 144.9, 144.1, 134.2, 130.1, 120.5, 120.4, 116.3, 116.2, 116.1, 113.5, 45.0, 43.0, 36.3, 31.7, 29.9, 24.1. HR-ESI-MS m/z: [M + H]^+^ calcd for C_19_H_23_O_4_ 315.1591, found 315.1588.
1-(3.4-Dihydroxyphenyl)-7-(4-hydroxyphenyl)heptan-3-one (8c)
Following the procedure as described for the preparation of compound 6a, the reaction of compound 7c (100 mg, 0.32 mmol) in EtOH (5 ml) and 10% Pd–C (10 mg) to give compound 8c (51 mg, 51%) as a brown solid. Melting point 90–93 °C. ^1^H NMR (Acetone-d6, 500 MHz): δ 8.08 (s, 1H), 7.71 (s, 1H), 7.67 (s, 1H), 6.99 (d, J = 8.5 Hz, 2H), 6.73 (d, J = 8.5 Hz, 2H), 6.70 (d, J = 8.0 Hz,1H), 6.68 (d, J = 2.0 Hz, 1H), 6.51 (dd, J = 8.0, 2.0 Hz,1H), 2.67 (m, 4H), 2.49 (m, 2H), 2.43 (m, 2H), 1.53 (m, 4H). ^13^C NMR (Acetone-d6, 125 MHz): δ 209.9, 156.4, 145.9, 144.1, 134.1, 134.0, 130.1, 120.4, 116.3, 116.0, 115.9, 45.0, 43.0, 35.5, 32.2, 29.9, 24.1. HR-ESI-MS m/z: [M + H]^+^ calcd for C_19_H_23_O_4_ 315.1591, found 315.1592.
4-(Methoxymethoxy)-1H-indole (10a)
To a solution of 4-hydroxyindole (100 mg, 0.75 mmol) in dry CH_2_Cl_2_ (10 ml) was added N,N-diisopropylethylamine (261 μl, 1.50 mmol) and chloromethyl methyl ether (MOMCl, 114 μl, 1.50 mmol) dropwise in an ice bath. The resultant was warmed to room temperature and stirred under N_2_ atmosphere for 2 h. The reaction mixture was diluted with distilled H_2_O (100 ml) and extracted with CH_2_Cl_2_ (3 50 ml). The combined organic layer was dried over anhydrous Na_2_SO_4_, filtered, and the solvent was concentrated in vacuo. The residue was purified over a silica gel column eluted with 11% EtOAc in n-hexane to give compound 10a (89 mg, 67%) as a light yellow oil. ^1^H NMR (CDCl_3_, 300 MHz): δ 8.17 (br, 1H), 7.08 (m, 3H), 6.76 (dd, J = 7.1, 1.4 Hz, 1H), 6.66 (t, J = 2.7 Hz, 1H), 5.34 (s, 2H), 3.54 (s, 3H). ESI-MS m/z: 178.1 [M + H]^+^.
6-Benzyloxy-1H-indole (10b)
To a solution of 6-hydroxyindole (2.50 g, 18.78 mmol) and K_2_CO_3_ (12.98 g, 93.88 mmol) in dimethylformamide (DMF) (100 ml) was stirred under N_2_ atmosphere for 1 h. The solution was added benzyl chloride (4.32 ml, 37.55 mmol) dropwise in an ice bath. The resultant was warmed to room temperature and stirred under N_2_ atmosphere for 13 h. The reaction mixture was diluted with distilled H_2_O (100 ml) and extracted with EtOAc (5 100 ml). The combined organic layer was dried over anhydrous Na_2_SO_4_, filtered, and the solvent was concentrated in vacuo. The residue was purified over a silica gel column eluted with 10% EtOAc in n-hexane to give compound 10b (3.31 g, 79%) as a yellow solid. Melting point 115–117 °C. ^1^H NMR (CDCl_3_, 300 MHz): δ 7.99 (br, 1H), 7.50 (d, J = 8.7 Hz, 1H), 7.45 (d, J = 6.9 Hz, 2H), 7.34 (m, 3H), 7.08 (dd, J = 3.2, 2.4 Hz, 1H), 6.93 (d, J = 2.2 Hz), 6.87 (dd, J = 8.7, 2.2 Hz, 1H), 6.47 (m, 1H), 5.10 (s, 2H). ESI-MS m/z: 224.11 [M + H]^+^.
6-(4-Methoxymethoxy-1H-indol-1-yl)hexan-2-one (11a)
To a solution of compound 10a (100 mg, 0.56 mmol) in DMF (5 ml) was added NaH (82 mg, 3.40 mmol). The resulting solution was stirred for 1.5 h. The reaction mixture was added 6-chloro-hexanone (332 μl, 2.51 mmol) dropwise and was stirred at room temperature under N_2_ atmosphere for 24 h. The reaction mixture was diluted with distilled H_2_O (100 ml) and extracted with EtOAc (3 50 ml). The combined organic layer was dried over anhydrous Na_2_SO_4_, filtered, and the solvent was concentrated in vacuo. The residue was purified over a silica gel column eluted with 11% EtOAc in n-hexane to give compound 11a (88 mg, 57%) as a brown oil. ^1^H NMR (CDCl_3_, 300 MHz): δ 7.09 (t, J = 8.0 Hz, 1H), 6.98 (m, 2H), 6.73 (d, J = 7.9 Hz, 1H), 6.57 (dd, J = 3.2, 0.7 Hz, 1H), 5.31 (s, 2H), 4.08 (t, J = 6.9 Hz, 2H), 3.52 (s, 3H), 2.38 (t, J = 7.2 Hz, 2H), 2.06 (s, 3H), 1.80 (m, 2H), 1.57 (m, 2H). ESI-MS m/z: 276.2 [M + H]^+^.
6-(6-Benzyloxy-1H-indol-1-yl)hexan-2-one (11b)
Following the procedure as described for the preparation of compound 11a, the reaction of compound 10b (100 mg, 0.45 mmol), NaH (43 mg, 1.79 mmol), 6-chloro-2-hexanone (176 μl, 1.33 mmol) in DMF (5 ml) gave compound 11b (55 mg, 38%) as a brown solid. Melting point 43–46 °C. ^1^H NMR (CDCl_3_, 300 MHz): δ 7.47 (m, 3H), 7.33 (m, 3H), 6.96 (d, J = 3.2 Hz, 1H), 6.84 (m, 2H), 6.39 (dd, J = 3.2, 0.5 Hz, 1H), 5.11 (s, 2H), 4.02 (t, J = 7.1 Hz, 2H), 2.37 (t, J = 7.1 Hz, 2H), 2.37 (s, 3H), 1.77 (m, 2H), 1.58 (m, 2H). ESI-MS m/z: 322.2 [M + H]^+^.
6-(4-Hydroxy-1H-indol-1-yl)hexan-2-one (12a)
To a solution of compound 11a (500 mg, 1.82 mmol) in MeOH (50 ml) at 0 °C was added 1 N HCl_(aq)_ (50 ml) dropwise. The resultant was warmed to room temperature and stirred under N_2_ for 22 h. The reaction mixture was diluted with distilled H_2_O (100 ml) and extracted with EtOAc (3 50 ml). The combined organic layer was dried over anhydrous Na_2_SO_4_, filtered, and the solvent was concentrated in vacuo. The residue was purified over a silica gel column eluted with 25% EtOAc in n-hexane to give compound 12a (121 mg, 29%) as a yellow solid. Melting point 91–94 °C. ^1^H NMR (CDCl_3_, 300 MHz): δ 7.04 (t, J = 8.0 Hz, 1H), 6.99 (d, J = 3.2 Hz, 1H), 6.91 (d, J = 8.3 Hz, 1H), 6.49 (m, 2H), 5.11 (s, 1H), 4.08 (t, J = 6.9 Hz, 2H), 2.39 (t, J = 7.2 Hz, 2H), 2.07 (s, 3H), 1.81 (m, 2H), 1.57 (m, 2H). ESI-MS m/z: 232.1 [M + H]^+^.
6-(6-Hydroxy-1H-indol-1-yl)hexan-2-one (12b)
To a solution of compound 11b (1.27 g, 3.95 mmol) and ammonium formate (2.49 g, 39.51 mmol) in MeOH (30 ml) was added catalysed 10% Pd–C (130 mg). The resultant was refluxed and stirred under N_2_ for 1.5 h. The reaction mixture was filtered through a celite pad. The residue was diluted with distilled H_2_O (100 ml) and extracted with EtOAc (3 50 ml). The combined organic layer was dried over anhydrous Na_2_SO_4_, filtered, and the solvent was concentrated in vacuo. The residue was purified over a silica gel column eluted with 28% EtOAc in n-hexane to give compound 12b (329 mg, 36%) as a white solid. Melting point 94–98 °C. ^1^H NMR (CDCl_3_, 300 MHz): δ 7.42 (d, J = 8.1 Hz, 1H), 6.93 (d, J = 3.1 Hz, 1H), 6.75 (d, J = 2.2 Hz, 1H), 6.65 (dd, J = 8.1, 2.2 Hz, 1H), 6.38 (dd, J = 3.1, 0.8 Hz, 1H), 6.65 (s, 1H), 3.99 (t, J = 6.9 Hz, 1H), 2.39 (t, J = 7.1 Hz, 1H), 2.07 (s, 3H), 1.77 (m, 2H), 1.57 (m, 2H). ESI-MS m/z: 232.1 [M + H]^+^.
(E)-1-(3,4-dihydroxyphenyl)-7-(4-hydroxy-1H-indol-1-yl)hept-1-en-3-one (13a)
Following the procedure as described for the preparation of compound 7a, the reaction of compound 12a (100 mg, 0.43 mmol) in THF (20 ml) with pyrrolidine (71 μl, 0.86 mmol), acetic acid (49 μl, 0.86 mmol), and 3,4- dihydroxybenzaldehyde (40 mg, 0.29 mmol) gave compound 13a (40 mg, 39%) as a yellow solid. Melting point 180–184 °C. ^1^H NMR (Acetone-d6, 300 MHz): δ 8.34 (br, 3H), 7.46 (d, J = 16.2 Hz, 1H), 7.16 (m, 2H), 7.04 (dd, J = 8.2, 2.1 Hz, 1H), 6.95 (m, 2H), 6.87 (d, J = 8.2 Hz, 1H), 6.58 (m, 2H), 6.45 (dd, J = 2.8, 5.4 Hz, 1H), 4.18 (t, J = 7.2 H, 2H), 2.67 (dd, J = 7.2 Hz, 2H), 1.87 (m, 2H), 1.64 (m, 2H). ^13^C NMR (Acetone-d6, 125 MHz): δ 199.6, 151.6, 148.7, 146.3, 143.1, 139.0, 127.9, 127.0, 124.4, 122.9, 122.7, 119.5, 116.3, 115.2, 104.1, 102.4, 98.6, 46.7, 40.2, 30.5, 22.3. HR-ESI-MS m/z: [M + H]^+^ calcd for C_21_H_22_O_4_N 352.1543, found 352.1535.
(E)-1-(3.4-dihydroxyphenyl)-7-(6-hydroxy-1H-indol-1-yl)hept-1-en-3-one (13b)
Following the procedure as described for the preparation of compound 7a, the reaction of compound 12b (50 mg, 0.22 mmol) in THF (5 ml) with pyrrolidine (24 μl, 0.29 mmol), acetic acid (17 μl, 0.29 mmol), and 3,4- dihydroxybenzaldehyde (19 mg, 0.14 mmol) gave compound 13b (27 mg, 55%) as a brown solid. Melting point 155–159 °C. ^1^H NMR (Acetone-d6, 300 MHz): δ 8.32 (br, 2H), 7.92 (br, 1H), 7.46 (d, J = 16.2 Hz, 1H), 7.34 (d, J = 8.5 Hz, 1H), 7.17 (d, J = 2.1 Hz, 1H), 7.08 (d, J = 3.2 Hz, 1H), 7.04 (dd, J = 8.2 Hz, 2.0 Hz, 1H), 6.86 (m, 2H), 6.65 (dd, J = 8.5, 2.1 Hz, 1H), 6.58 (dd, J = 16.2 Hz, 1H), 6.31 (dd, J = 3.1, 0.6 Hz, 1H), 4.10 (t, J = 7.0 Hz, 2H), 2.67 (t. J = 7.3 Hz, 2H), 1.84 (m, 2H), 1.64 (m, 2H). ^13^C NMR (Acetone-d6, 125 MHz): δ 199.6, 154.1, 148.7, 146.3, 143.2, 138.0, 127.9, 127.3, 124.4, 123.3, 122.7, 121.8, 116.4, 115.2, 110.2, 101.4, 95.8, 46.4, 40.3, 30.4, 22.4. HR-ESI-MS m/z: [M + H]^+^ calcd for C_21_H_22_O_4_N 352.1543, found 352.1537.
1-(3,4-Dihydroxyphenyl)-7-(4-hydroxy-1H-indol-1-yl)heptan-3-one (14a)
Following the procedure as described for the preparation of compound 6a, the reaction of compound 13a (50 mg, 0.14 mmol) in EtOAc (10 ml) with 10% Pd–C (5 mg) gave compound 14a (37 mg, 74%) as a black solid. ^1^H NMR (Acetone-d6, 300 MHz): δ 8.30 (s, 1H), 7.69 (s, 2H), 7.12 (d, J = 3.2 Hz, 1H), 6.94 (m, 2H), 6.70 (d, J = 8.0 Hz, 1H), 6.67 (d, J = 2.1 Hz,1H), 6.55 (d, J = 3.2 Hz, 1H), 6.50 (dd, J = 8.0, 2.1 Hz, 1H), 6.45 (dd, J = 7.0, 1.0 Hz, 1H), 4.12 (t, J = 7.0 Hz, 2H), 2.64 (m, 4H), 2.43 (t, J = 7.2 Hz, 2H), 1.77 (m, 2H), 1.52 (m, 2H). ^13^C NMR (Acetone-d6, 125 MHz): δ 209.6, 151.6, 145.7, 144.0, 139.0, 134.0, 127.0, 123.0, 120.2, 119.5, 116.2, 115.9, 104.1, 102.4, 98.6, 46.7, 44.9, 42.4, 30.4, 29.8, 21.6. HR-ESI-MS m/z: [M + H]^+^ calcd for C_21_H_24_O_4_N 354.1700, found 354.1696.
1-(3,4-Dihydroxyphenyl)-7-(6-hydroxy-1H-indol-1-yl)heptan-3-one (14b)
Following the procedure as described for the preparation of compound 6a, the reaction of compound 13b (68 mg, 0.19 mmol) in MeOH (5 ml) with 10% Pd–C (7 mg) gave compound 14b (40 mg, 58%) as a brown oil. ^1^H NMR (Acetone-d6, 300 MHz): δ 7.89 (s, 1H), 7.66 (s, 1H), 7.63 (s, 1H), 7.33 (d, J = 8.5 Hz, 1H), 7.05 (d, 3.2 Hz, 1H), 6.81 (d, J = 2.1 Hz, 1H), 6.70 (d, J = 8.0 Hz, 1H), 6.67 (d, J = 2.1 Hz, 1H), 6.63 (dd, J = 8.5, 2.1 Hz, 1H), 6.50 (dd, J = 8.0, 2.1 Hz, 1H), 6.29 (dd, J = 3.2, 0.7 Hz, 1H), 4.06 (t, J = 7.0 Hz, 2H), 2.65 (m, 4H), 2.43 (t, J = 7.2 Hz, 2H), 1.76 (m, 2H), 1.53 (m, 2H). ^13^C NMR (Acetone-d6, 125 MHz): δ 209.6, 154.1, 145.7, 144.0, 138.0, 134.0, 127.3, 123.3, 121.8, 120.2, 116.2, 116.0, 110.2, 101.4, 95.8, 46.4, 44.9, 42.5, 21.6. HR-ESI-MS m/z: [M + H]^+^ calcd for C_21_H_24_O_4_N 354.1700, found 354.1695.
(E)-1-(3,4-dihydroxyphenyl)-7-(4-(methoxymethoxy)-1H-indol-1-yl)hept-1-en-3-one (15)
Following the procedure as described for the preparation of compound 7a, the reaction of compound 11a (880 mg, 3.20 mmol) in THF (90 ml) with pyrrolidine (350 μl, 4.26 mmol), acetic acid (243 μl, 4.26 mmol), and 3,4-dihydroxybenzaldehyde (294 mg, 2.13 mmol) gave compound 15 (440 mg, 52%) as a yellow solid. Melting point 129–132 °C. ^1^H NMR (Acetone-d6, 300 MHz): δ 8.29 (br, 2H), 7.45 (d, J = 16.2 Hz,1H), 7.22 (d, J = 3.2 Hz, 1H), 7.15 (d, J = 2.1 Hz, 1H), 7.12 (m, 1H), 7.04 (m, 2H), 6.86 (d, J = 8.2 Hz, 1H), 6.69 (dd, J = 7.6, 0.8 Hz, 1H), 6.58 (d, J = 16.2 Hz, 1H), 6.51 (dd, J = 3.2, 0.7 Hz, 1H), 5.30 (s, 2H), 4.22 (t, J = 7.0 Hz, 2H), 3.47 (s, 3H), 2.68 (t, J = 7.3 Hz, 2H), 1.88 (m, 2H), 1.64 (m, 2H). ^13^C NMR (Acetone-d6, 125 MHz): δ 199.6, 151.7, 148.7, 146.3, 143.1, 138.7, 127.9, 127.7, 124.4, 122.7, 122.6, 120.9, 116.4, 115.2, 104.8, 104.0, 98.7, 95.4, 56.0, 46.7, 40.2, 30.6, 22.3. HR-ESI-MS m/z [M + H]^+^ calcd for C_23_H_26_O_5_N 396.1805, found 396.1978.
6-(1H-indol-1-yl)hexan-2-one (17)
Following the procedure as described for the preparation of compound 11a, the reaction of indole (100 mg, 0.85 mmol) and NaH (82 mg, 3.42 mmol) in DMF (5 ml) with 6-chloro-2-hexanone (332 μl, 2.55 mmol) gave compound 17 (93 mg, 51%) as a yellow oil. ^1^H NMR (CDCl_3_, 300 MHz): δ 7.61 (m, 1H), 7.32 (dd, J = 8.2, 0.8 Hz, 1H), 7.19 (m, 1H), 7.08 (m, 2H), 6.47 (dd, J = 3.1, 0.8 Hz, 1H), 4.11 (t, J = 6.9 Hz, 2H), 2.39 (t, J = 7.2 Hz, 2H), 1.82 (m, 2H), 1.57 (m, 2H). ESI-MS m/z: 216.1 [M + H]^+^.
(E)-1-(3,4-dihydroxyphenyl)-7-(1H-indol-1-yl)hept-1-en-3-one (18)
Following the procedure as described for the preparation of compound 7a, the reaction of compound 17 (100 mg, 0.46 mmol) in THF (10 ml), pyrrolidine (50 μl, 0.61 mmol), acetic acid (35 μl, 0.61 mmol), and 3,4-dihydroxybenzaldehyde (43 mg, 0.31 mmol) gave compound 18 (81 mg, 78%) as a orange solid. Melting point 165–169 °C. ^1^H NMR (DMSO-d6, 300 MHz): δ 9.28 (br, 2H), 7.53 (d, J = 7.8 Hz, 1H), 7.47 (d, J = 8.1 Hz, 1H), 7.41 (d, J = 16.2 Hz, 1H), 7.37 (d, J = 3.2 Hz, 1H), 7.11 (m, 1H), 7.05 (d, J = 2.0 Hz, 1H), 6.99 (m, 2H), 6.76 (d, J = 8.1 Hz, 1H), 6.51 (d, J = 16.2 Hz, 1H), 6.41 (dd, J = 3.2, 0.6 Hz, 1H), 4.19 (t, J = 6.9 Hz, 2H), 2.65 (t, J = 7.1 Hz, 2H), 1.77 (m, 2H), 1.50 (m, 2H). ^13^C NMR (Acetone-d6, 125 MHz): δ 199.5, 148.7, 146.3, 143.1, 137.0, 129.7, 128.9, 127.9, 124.4, 122.7, 121.8, 121.4, 119.7, 116.4, 115.2, 110.4, 101.4, 46.5, 40.2, 30.6, 22.3. HR-ESI-MS m/z: [M + H]^+^ calcd for C_21_H_22_O_3_N 336.1594, found 336.1587.
1-(3,4-Dihydroxyphenyl)-7-(1H-indol-1-yl)heptan-3-one (19)
Following the procedure as described for the preparation of compound 6a, the reaction of compound 18 (226 mg, 0.67 mmol) in EtOAc (25 ml) with 10% Pd–C (23 mg) gave compound 19 (120 mg, 53%) as a white solid. Melting point 110–113 °C. ^1^H NMR (Acetone-d6, 300 MHz): δ 7.66 (s, 1H), 7.63 (s, 1H), 7.54 (m, 1H), 7.42 (dd, J = 8.2, 0.8 Hz, 1H), 7.26 (d, J = 3.2 Hz, 1H), 7.13 (ddd, J = 8.2, 7.2, 1.1 Hz, 1H), 7.00 (ddd, J = 7.9, 7.0, 0.8 Hz, 1H), 6.70 (d, J = 8.1 Hz, 1H), 6.66 (d, J = 2.1 Hz, 1H), 6.50 (dd, J = 7.9, 2.1 Hz, 1H), 6.42 (dd, J = 3.2, 0.8 Hz, 1H), 4.19 (t, J = 7.0 Hz, 2H), 2.65 (m, 4H), 2.44 (t, J = 7.2 Hz, 2H), 1.80 (m, 2H), 1.53 (m, 2H). ^13^C NMR (Acetone-d6, 125 MHz): δ 209.5, 145.7, 144.0, 137.0, 134.0, 129.6, 128.9, 121.9, 121.4, 120.2, 119.7, 116.2, 115.9, 110.3, 46.5, 44.7, 42.4, 30.4, 29.8, 21.6. HR-ESI-MS m/z: [M + H]^+^ calcd. for C_21_H_24_O_3_N, 338.1751, found 338.1744.
Molecular docking analysis
The docking software LeadIT40 (version 2.3.2) was utilised to analyse the interactions between the compounds and LIMK1 (PDB ID: 3S95). The co-crystal structure of LIMK1 was obtained from the Protein Data Bank (PDB). The location of the co-crystallised ligand was used to define the binding site, which was defined with a radius of 10 Å. Water molecules were removed from the binding site prior to analysis. All docked compounds were protonated in an aqueous solution. The FlexX docking module of LeadIT was employed to conduct the docking analysis. Ligand placement is guided by a hybrid enthalpy–entropy approach, ensuring that both energetic and entropic contributions are considered when evaluating binding interactions. In the scoring section, access scaling is enabled, applying uniform settings to all hydrogen bond–relevant contact types. The thresholds are set so that contacts contribute fully at 0.30 and contribute no score at 0.70 or higher. For clash handling, the system allows a maximum protein–ligand overlap volume of 2.9 ų, indicating a moderate tolerance, while intra-ligand clashes are controlled with a clash factor of 0.6. Hydrogens are included in internal clash evaluations for greater accuracy. Finally, the docking details specify that up to 200 solutions per iteration and 200 solutions per fragmentation will be generated, which provides a relatively broad sampling of conformational space. Overall, this configuration balances flexibility with efficiency, applying moderate clash stringency while enabling extensive exploration of possible ligand poses.
Kinase inhibition assay
The inhibitory activities of the compounds against kinases were tested using the SelectScreen Kinase assay from Thermo Fisher Scientific in Waltham, MA, USA. The LanthaScreen Eu kinase binding assay was used to validate the LIMK1 inhibition activity of the compounds, and was performed according to the manufacturer’s protocol (https://www.thermofisher.com/tw/zt/home/industrial/pharma-biopharma/drug-discovery-development/target-and-lead-identification-and-validation/kinasebiology/kinase-activity-assays/lanthascreentm-eu-kinase-binding-assay/lanthascreen-eu-kinase-binding-assay-validation-table.html). This endpoint assay quantifies inhibitor binding through the displacement of a fluorescently labelled ATP-competitive tracer from LIMK1. A mixture of the 100× compounds, 2× LIMK1/antibody mixture (Eu-anti-His), 4× tracer 236, and kinase buffer were mixed sequentially. After incubation at room temperature for 1 h, the mixture was analysed using a fluorescence plate reader. The kinase inhibition assay results were expressed as the average of two replicates.
Kinase panel screening was carried out using the SelectScreen Kinase Assay (Thermo Fisher Scientific, Waltham, MA, USA). Three complementary assay formats were applied: the Z′-LYTE kinase assay, LanthaScreen Eu kinase binding assay, and Adapta Universal kinase assay. The kinases selected for this study and the corresponding assay formats are summarised in Supplementary Table S1.
The Z′-LYTE Kinase Assay (Thermo Fisher Scientific, Waltham, MA, USA) was used to evaluate selected kinases’ activity, according to the manufacturer’s instructions (https://www.thermofisher.com/tw/zt/home/industrial/pharma-biopharma/drug-discovery-development/target-and-lead-identification-and-validation/kinasebiology/kinase-activity-assays/z-lyte/z-lyte-reactivity-table.html). This endpoint assay quantifies kinase activity by detecting peptide substrate phosphorylation. Briefly, a 2× mixture of selected kinase and relevant substrate was prepared in kinase buffer (0.01% polyoxyethylene lauryl ether (BRIJ-35), 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.5, 1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), and 10 mM MgCl_2_). After 1 h incubation at room temperature, 5 μl of 1:128 diluted development reagent solution was added, followed by an additional 1 h incubation. Fluorescence was recorded using a multimode plate reader.
The Adapta Universal Kinase Assay (Thermo Fisher Scientific, Waltham, MA, USA) was used to assess kinase activity. This homogeneous, fluorescence-based immunoassay quantifies kinase activity by detecting ADP produced during the enzymatic reaction, according to the manufacturer’s instructions (https://www.thermofisher.com/tw/zt/home/industrial/pharma-biopharma/drug-discovery-development/target-and-lead-identification-and-validation/kinasebiology/kinase-activity-assays/adapta-universal-kinase-assay.html).
Cell culture
Colorectal cell line, HCT-116, was purchased from the Bioresource Collection and Research Centre (BCRC, Hsinchu, Taiwan), while HT-29 was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Both cell lines were maintained in McCoy’s 5 A medium (with L-glutamine and 2.2 g/L NaHCO_3_, M4892, Sigma) supplemented with 10% foetal bovine serum (Corning Incorporated, NY, USA) and 1% penicillin–streptomycin–amphotericin B (100 unit/ml penicillin G, 0.1 mg/ml streptomycin sulphate, 0.25 μg/ml amphotericin B, Corning Incorporated, NY, USA). All cultures were maintained in a humidified incubator at 37 °C with 5% CO_2_.
MTT assay
Cells were seeded in 96-well plates at a density of 3 10^3^ cells/well in 100 μl of culture medium and treated with compounds at the indicated concentrations (0.3, 1, 3, 10, and 30 μM) for 72 h. Following treatment, the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (5 mg/ml in Phosphate-buffered saline (PBS)) was added to each well to reach a final concentration of 0.5 mg/ml, and the cells were incubated at 37 °C for 1 h. The resulting formazan crystal was dissolved in 100 µl of DMSO, and the absorbance was measured at 550 nm using an ELISA reader (Molecular Device, Sunnyvale, CA, USA).
Sulforhodamine B assay
Cells were seeded in 96-well plates at a density of 3 10^3^ cells/well in 100 μl of culture medium and treated with compounds at the indicated concentrations for 72 h. After treatment, cells were fixed with 10% trichloroacetic acid for 20 min and washed with H_2_O. The cells were then stained with 0.4% sulforhodamine B (SRB) in 1% acetic acid for 20 min, followed by two washes with 1% acetic acid. After the well-dried, the protein-bound dye was dissolved in 100 µl of 10 mM Tris base solution, and the absorbance was measured at 515 nm using an ELISA reader (Molecular Device, Sunnyvale, CA, USA).
Analysis of cell cycle by flow cytometry
Cells were seeded in six-well plates at a density of 5 10^5^ cells/well in a 2 ml culture medium and treated with compounds at the indicated concentration for 72 h. After treatment, cells were collected and fixed with 75% (v/v) ice-cold ethanol at −20 °C for 20 min. The cells were then centrifuged to remove the buffer, resuspended, and stained with 0.5 ml of propidium iodide (PI) staining buffer (80 µg/ml PI, 100 µl/ml RNase A, and 1% Triton X-100 in PBS). The cell cycle distribution was analysed using a BD Accuri™ Flow cytometer and software (Becton Dickinson, Mountain View, CA, USA).
Statistical analysis
All biological experiments were repeated at least three times to ensure reproducibility and reliability. Data are presented as the mean ± standard deviation (SD) or expressed as a percentage of the control group, depending on the nature of the experiment. Statistical analyses were performed using a one-way analysis of variance (non-parametric test) in GraphPad Prism. A p values of <0.05 was considered statistically significant.
Result and discussion
Design rationale
A previous study found that curcumin has LIMK inhibitory activity37. To better understand the interactions between curcumin and the LIMK1 binding site, we conducted a molecular docking analysis. The docking results showed that curcumin occupies the binding site (Figure 2(A)). Specifically, the 4-hydroxy-3-methoxyphenyl group of curcumin acts as a hinge binder, forming two hydrogen bonds with hinge residues E414 and I416. In addition, the aromatic ring of curcumin interacts with the LIMK1 binding site through π-alkyl interactions with residues L345, V366, and L467. Two aromatic rings of curcumin are attached together through a seven-carbon length and extend into another pocket of the binding site. This group contributes an additional hydrogen bond with residue G351. These observed interactions suggest that the inhibitory activity of curcumin is due to its interaction with LIMK1, indicating diarylheptanoid as a promising scaffold for a LIMK1 inhibitor.
Interaction analysis of curcumin, compounds I and III–VI with LIMK1 binding site. Docking poses of (A) curcumin (yellow) and (B) compound I (salmon) in the LIMK1 binding site (PDB ID: 3S95). (C) Superimposed docking poses of compounds I, III (lime-green), IV (blue-green), V (magenta), and VI (orange). (D) Docking pose of compound I superimposed onto the pose of compound IV in LIMK1 (surface model). Hydrogen bonds are represented as green dashed lines.
Compound I can be classified as a diarylheptanoid, which was designed and synthesised in our previous research41. The catechol group of compound I might act as a hydrogen bond donor or acceptor simultaneously, which is advantageous for interacting with hinge residues. Consequently, compound I was evaluated for its ability to inhibit LIMK1, and the result indicated stronger activity compared to curcumin, with an IC_50_ value of 1.59 µM (Table 1). The docking result revealed that compound I forms more hydrogen bonds with hinge residues than curcumin, primarily due to the presence of the catechol group (Figure 2(B)). Specifically, compound I forms an additional hydrogen bond with hinge residue V366 through the 3-hydroxy group (Figure 2(B)), which may explain its superior LIMK1 inhibition compared to curcumin. Therefore, the catechol has the potential to serve as a hinge binder in the design of a new series of diarylheptanoid-based LIMK1 inhibitors.
To further investigate the impact of the α-β saturation on LIMK1 inhibitory activity, compound II, which has a saturated linker, was compared to compound I. The result showed that saturated compound II only exhibited 30% inhibition at 10 μM (Table 1), suggesting the importance of the unsaturated bond for LIMK1 inhibition. Subsequently, unsaturated compounds III–VI were evaluated for their LIMK1 inhibition at 10 μM to study the effects of carbon chain length. The experimental results revealed that compounds with shorter carbon chains did not demonstrate better LIMK1 inhibition than compound I (Table 1). Docking results of the compounds indicated similar interactions in the hinge region (Figure 2(C)). However, their B ring contributed to different interactions with LIMK1 residues due to varying linker lengths. Among these compounds, compound IV showed less potency in LIMK1 inhibition. The docking pose of compound IV in LIMK1 was compared to that of compound I (Figure 2(D)). It was found that the chain shortening of compound III resulted in its B ring not being able to extend into another pocket containing residue G351, possibly accounting for its weaker potency.
In short, we found that catechol-containing diarylheptanoid is a suitable scaffold with stronger inhibitory activity than curcumin. To enhance the inhibitory activity, we selected compound I as a lead compound and synthesised a series of catechol-containing diarylheptanoid derivatives. Specifically, we added a hydroxy group to the B ring to create compounds 7a–c and 8a–c, aiming to form additional hydrogen bonds. Moreover, to achieve extra π interaction with LIMK1, we developed compounds 13a, 13b, 14a, 14b, 15, 18, and 19 by replacing the B ring with indole. These changes in structure are expected to enhance the LIMK1 inhibitory activity.
Chemistry
Synthesis of compounds 7a–c and 8a–c
Compounds 7a–c and 8a–c were synthesised as shown in Scheme 1. Initially, the Horner–Wadsworth–Emmons reaction of benzyl-protected benzaldehydes 1a–c with triethyl phosphonoacetate in the presence of NaH provided compounds 2a–c, respectively. Reduction of compounds 2a–c with DIBAL-H generated corresponding alcohols 3a–c. Oxidation of compounds 3a–c using MnO_2_ produced aldehydes 4a–c, respectively. Wittig reaction of compounds 4a–c with 1-(triphenylphosphoranylidene)-1-propanone yielded compounds 5a–c. Atmospheric hydrogenation of compounds 5a–c using catalytic 10% Pd–C removed the benzyl protecting group, producing the corresponding compounds 6a–c. Aldol condensation of compounds 6a–c with 3, 4-dihydroxybenzaldehyde generated compounds 7a–c. Repeated hydrogenation of compounds 7a–c gave saturated compounds 8a–c, respectively.
Reagents and conditions: (a) Triethyl phosphonoacetate, NaH, dry THF, 0 °C; (b) DIBAL-H, dry CH2Cl2, −30 °C; (c) MnO2, toluene, 110 °C; (d) 1-Triphenylphosphoranylidene-2-propanone, dry toluene, 110 °C; (e) 10% Pd–C, H2, EtOH, RT; and (f) 3, 4-dihydroxybenzaldehyde, pyrrolidine, AcOH, THF, 66 °C.
Synthesis of compounds 13a, 13b, 14a, 14b, 15, 18, and 19
Compounds 13a, 13b, 14a, 14b, 15, 18, and 19 were synthesised as outlined in Scheme 2. The hydroxy groups of indoles 9a and 9b were protected by reacting with chloromethyl methyl ether (MOMCl) and benzyl chloride (BnCl) to produce compounds 10a and 10b, respectively. Subsequent nucleophilic substitution of compounds 10a and 10b with 6-chloro-2-hexanone generated corresponding compounds 11a and 11b. Deprotection of compounds 11a and 11b yielded compounds 12a and 12b, which then underwent aldol condensation with 3,4-dihydroxybenzaldehyde to produce compounds 13a and 13b. Atmospheric hydrogenation of compounds 13a and 13b yielded compounds 14a and 14b, respectively. Repeated aldol condensation of compound 11a afforded compound 15. The synthetic approach for compound 19 was similar to that for compounds 14a and 14b. All compounds tested for inhibitory activity were further confirmed for their molecular composition by high-resolution mass spectrometry. Detailed characterisation spectra (^1^H and ^13^C NMR) for the synthesised compounds are provided in both the “Materials and Methods” section and the Supplementary Materials.
Reagents and conditions: (a) for 10a: MOMCl, DIPEA, CH2Cl2, RT; for 10b: BnCl, K2CO3, DMF, 80 °C; (b) 6-chloro-2-hexanone, NaH, DMF, RT; (c) for 12a: 1 N HCl(aq), MeOH, RT; for 12b: 10% Pd–C, NH4HCO2, MeOH, 80 °C; (d) 3,4-dihydroxybenzaldehyde, pyrrolidine, acetic acid, THF, RT; and (e) 10% Pd–C, H2, EtOAc, RT.
Biological evaluation
Inhibition of compounds 7a–c, 8a–c, 13a, 13b, 14a, 14b, 15, 18, and 19 against LIMK1
The synthesised compounds were evaluated for their ability to inhibit LIMK1. The percentage of LIMK1 inhibition for each compound was measured at a concentration of 10 μM (Table 2). Compounds that exhibited over 80% inhibitory activity at this concentration were chosen for further evaluation of their IC_50_ values. Similar to the previously mentioned results, α-β unsaturated compounds 7a–c, 13a, 13b, and 18 showed greater potency compared to the saturated compounds 8a–c, 14a, 14b, and 19. With compounds 7a–c, compound 7c, which had a 4′-hydroxy substitution, displayed higher LIMK1 inhibition compared to compounds with 3′ and 2′-hydroxy substitution. The docking result suggested that the improvement may be attributed to the formation of an additional hydrogen bond between residue G351 and the 4′-hydroxy group of compound 7c (Figure 3(A)). In the compounds containing an indole group, compound 13a with a 4′-hydroxy group showed better LIMK1 inhibitory activity than compound 13b with a 6′-hydroxy group. The 4′-hydroxy group of compound 13a forms a hydrogen bond with residue F350 (Figure 3(B)). Compounds 15 and 18, which lacked a hydroxy group on the indole, exhibited lower potency than compound 13a. Additionally, among these compounds tested, compound 13a demonstrated the most potent LIMK1 inhibition. These results also suggested that the incorporation of an additional hydroxy group in the B ring/indole and the substitution of the B ring with indole enhance the LIMK1 inhibitory activity of the compounds.
Molecular docking analysis of compounds 7c, 13a, and XV in LIMK1. Docking poses of (A) compound 7c (slate), (B) compound 13a (pink), and (C) compound XV (yellow) with LIMK1 (grey). The residues involved in the interaction are labelled as depicted. Hydrogen bonds are depicted as green dashed lines.
Inhibition of compounds VII–XV against LIMK1
The impact of the hydroxy group on A ring against LIMK1 was investigated using nine previously synthesised compounds VII–XV (Table 3). Compound VII, which was without hydroxy groups, and compounds VIII–IX, which had only one hydroxy group, lost their inhibitory activity. Additionally, compounds with an electron-withdrawing group at the 3-position (compounds X–XII) or an electron-donating group (compound XIII) exhibited reduced inhibitory activity. However, compound XV, which consists of a pyrogallol group, showed the most potent LIMK1 inhibition with an IC_50_ value of 569 nM. Similar to the catechol group of compound I, compound XV also formed the same three hydrogen bonds with hinge residues E414 and I416 (Figure 3(B)). Additionally, the 3,4,5-trihydroxy group of compound XV established an extra hydrogen bond with residue I416. Furthermore, compound XV formed another hydrogen bond with residue T413 near the hinge region. In total, five hydrogen bonds were formed between compound XV and LIMK1. The polar interaction allows compound XV to bind with LIMK1 and stabilise it. These findings suggest that having at least two hydroxy groups on the A ring is essential for LIMK1 inhibition. The above results provide valuable information for constructing SAR.
To elucidate the effect of saturated and unsaturated compounds, compound S was designed to compare with compound I. As shown in Supplementary Figure S48(A), both compounds share a common caffeoyl moiety (Group A), whereas Group B differs by the presence of an unsaturated chain in compound S and a saturated chain in compound I. The 2D interaction mapping (Supplementary Figure S48(B)) revealed that Group A consistently forms hydrogen bonds with residues E414 and I416. These interactions suggest that the caffeoyl group functions as a conserved anchoring pharmacophore responsible for stable binding within the LIMK1 binding site.
Superimposed docking poses (Supplementary Figure S48(C)) further demonstrated that both compounds adopt similar orientations in Group A, whereas the structural divergence of Group B leads to distinct conformational adaptations. The unsaturated chain of compound S is positioned in closer proximity to the hydrophobic cavity near G351, which may promote additional interactions. In contrast, the saturated chain of compound I provides greater conformational flexibility. Taken together, these findings indicate that while Group A ensures a conserved interaction motif across both compounds, Group B critically modulates binding affinity.
Kinase selectivity of compound 13a
To evaluate the kinase selectivity of compound 13a, a panel of 40 kinases from various families was selected to determine its inhibitory selectivity further (Figure 4). The inhibition percentages for compound 13a across these kinases were all below 50% at a concentration of 1 μM. Specifically, it exhibited 44%, 32%, and 30% inhibition percentages for WEE1, ICK, and LIMK2, respectively. Notably, compound 13a demonstrated lower potency in inhibiting LIMK2 compared to LIMK1. A decrease in LIMK2 levels results in an increased accumulation of β-catenin in the nucleus, which activates the Wnt signalling pathway9. This activation is crucial for advancing CRC progression9. Therefore, the selective inhibition of LIMK1 over LIMK2 emphasises the advantages of compound 13a. Besides that, LIMK1 belongs to the tyrosine kinase-like (TKL) kinase family, and compound 13a displayed selectivity for other TKL family, including IRAK4, MLK1, BRAF, and TGFBR1. Given the high homology among the catalytic domains of kinases, most ATP-binding inhibitors can unintentionally affect multiple kinases, potentially leading to unwanted side effects. Fortunately, the selectivity analysis results indicate that compound 13a shows a preference for inhibiting LIMK1 (Figure 4).
Inhibition activity of compound 13a against a panel of representative kinases.
The pharmacokinetic and drug-likeness properties of compound 13a were evaluated using the ADMETlab 3.042 platform. As summarised in Supplementary Table S2, compound 13a displays physicochemical, absorption, distribution, metabolism, and excretion (ADME) profiles characteristic of drug-like small molecules. Compound 13a possesses a molecular weight of 351.15, a logP value of 2.95, and a topological polar surface area (TPSA) of 82.69 Å^2^, all within the optimal range for small-molecule drugs, suggesting a favourable balance between membrane permeability and aqueous solubility. Furthermore, compound 13a complies with multiple medicinal chemistry filters, including Lipinski’s Rule of Five, Pfizer, GSK, and Golden Triangle criteria, collectively supporting its overall drug-likeness.
Regarding ADME (Supplementary Table S2), the predicted permeability profiles revealed that the Caco-2 permeability (−5.091) was within the acceptable range for permeable compounds, and the compound is predicted to be a non-substrate of P-glycoprotein (0.02), suggesting that its cellular uptake is unlikely to be restricted by efflux transporters. Predicted human intestinal absorption (0.038) also indicates possible oral absorption. Additionally, compound 13a demonstrates high plasma protein binding (95.385%), low blood–brain barrier (BBB) penetration (0.02), and acceptable liver microsomal stability (0.634). The predicted plasma clearance (CL_plasma_ = 8.664 ml/min/kg) indicates moderate clearance, while the half-life (T1/2 = 0.932 h) classifies it as a short half-life compound. Although the compound shows inhibitory potential towards CYP1A2 (0.983), CYP3A4 (0.791), and CYP2B6 (1.0), no broad CYP promiscuity is predicted, minimising the likelihood of multi-pathway interference.
To further evaluate the safety profile of compound 13a, the toxicity was predicted using ProTox-III21^,^43. ProTox-III classifies compounds into six categories according to their predicted median lethal dose (LD_50_), where class one represents the most lethal and class six the safest. The analysis placed compound 13a in class 4 with a predicted LD_50_ of 1000 mg/kg (Supplementary Figures S1–S48). Collectively, these results indicate that compound 13a possesses balanced drug-like properties and an acceptable predicted safety margin, with only minor immunotoxicity and respiratory alerts, supporting its potential suitability for further development.
Cytotoxicity evaluation of compounds 13a and XV against CRC cells
LIMK1 is overexpressed and showed higher activity in CRC20. LIMK1 is related to cancer cell proliferation, migration, invasion, and metastasis. It has been proved that overexpressed LIMK1 in CRC promotes tumour growth and progression, while downregulated LIMK1 inhibits the growth of CRC cells in vitro and in vivo20. Among these compounds, compounds 13a and XV showed the most effective LIMK1 inhibition. To understand the effect of compounds 13a and XV in CRC cell growth, compounds 13a and XV were further evaluated using MTT assay and SRB assay (Table 4). MTT assay is used to measure the cell metabolic activity, while SRB assay is used to assess the protein content. Both assays can be used to evaluate the effects of compounds on cell viability and proliferation44. The results indicated that compound 13a inhibited the metabolic activity of both HCT-116 and HT-29 cells, exhibiting IC_50_ values of 18.24 and 28.78 µM, respectively, as determined by the MTT assay (Figure 5). Additionally, the SRB assay showed that compound 13a suppressed the growth of HCT-116 and HT-29 cells, with GI_50_ values of 14.65 and 10.36 µM, respectively. In contrast, compound XV did not inhibit the growth of HCT-116 cells at a concentration of 30 µM and showed reduced potency in HT-29 cells. These results suggest that compound 13a possesses a potent anti-proliferative effect on HCT-116 and HT-29 CRC cells.
*The anticancer effect of LIMK1 inhibitors. Cancer cells were treated with compound 13a at indicated concentrations (0.3, 1, 3, 10, and 30 μM) for 72 h in (A) HCT-116 and (B) HT-29 cells. The cell viability and cell proliferation were detected by MTT and SRB assays, respectively. All results present mean ± SD of three independent experiments. *p < 0.05 and *p < 0.01 compared to control group (ctrl.).
Cell cycle distribution of compound 13a
To reveal the effect of compound 13a treatment on the cell cycle, the cell cycle distribution of HCT116 and HT-29 cells was determined after treatment with various concentrations of compound 13a for 72 h. The results for HCT116 cells indicated a dose-dependent increase in the S phase and a decrease in the G0/G1 phase (Figure 6). Previous research indicated that LIMK1 expression in prostatic hyperplasia cells caused a transient G1/S phase arrest and delayed G2/M progression45. Thus, the effects of compound 13a on the cell cycle might be attributed to its inhibition of LIMK1. In both cell lines, the population of Sub-G1 cells increased in a dose-dependent manner following treatment with compound 13a, which suggests that compound 13a induced apoptosis. Accordingly, this result supported that compound 13a showed anti-proliferative and cytotoxic effects due to LIMK1 inhibition.
*Compound 13a induced cancer cell apoptosis. (A) HCT-116 and (B) HT-29 cells were treated with compound 13a with 1, 3, 10, and 30 μM for 72 h. The quantitative analysis of cell cycle distribution and percentage of sub-G1 population were analysed by flow cytometry. *p < 0.01 compared to control group (ctrl.).
Conclusion
In conclusion, we have designed and synthesised a novel series of compounds featuring a diarylheptanoid scaffold and conducted an extensive exploration of the LIMK1 inhibitory activity to investigate the SAR. Compound XV showed the best LIMK1 inhibitory activity. Meanwhile, compound 13a demonstrated greater cytotoxicity in both HCT-116 and HT-29 cells than compound XV. Treatment with compound 13a in HCT-116 cells led to an increase in the S phase and a decrease in the G0/G1 phase in a dose-dependent manner, like previous LIMK1 inhibitors. These results suggest that compound 13a has potent and selective LIMK1 inhibitory activity and induces apoptosis in HCT-116 and HT-29 cells, making it a promising lead compound for further optimisation.
Supplementary Material
Supplementaory material 20251022.docx
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Scott RW, Olson MF. Lim kinases: function, regulation and association with human disease. J Mol Med. 2007;85(6):555–568.17294230 10.1007/s 00109-007-0165-6 · doi ↗ · pubmed ↗
- 2Manetti F. Lim kinases are attractive targets with many macromolecular partners and only a few small molecule regulators. Med Res Rev. 2012;32(5):968–998.22886629 10.1002/med.20230 · doi ↗ · pubmed ↗
- 3Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by lim-kinase. Nature. 1998;393(6687):805–809.9655397 10.1038/31729 · doi ↗ · pubmed ↗
- 4Mizuno K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal. 2013;25(2):457–469.23153585 10.1016/j.cellsig.2012.11.001 · doi ↗ · pubmed ↗
- 5Ghosh M, Song X, Mouneimne G, Sidani M, Lawrence DS, Condeelis JS. Cofilin promotes actin polymerization and defines the direction of cell motility. Science. 2004;304(5671):743–746.15118165 10.1126/science.1094561 · doi ↗ · pubmed ↗
- 6Sumi T, Matsumoto K, Takai Y, Nakamura T. Cofilin phosphorylation and actin cytoskeletal dynamics regulated by rho- and cdc 42-activated lim-kinase 2. J Cell Biol. 1999;147(7):1519–1532.10613909 10.1083/jcb.147.7.1519 PMC 2174243 · doi ↗ · pubmed ↗
- 7Agnew BJ, Minamide LS, Bamburg JR. Reactivation of phosphorylated actin depolymerizing factor and identification of the regulatory site. J Biol Chem. 1995;270(29):17582–17587.7615564 10.1074/jbc.270.29.17582 · doi ↗ · pubmed ↗
- 8Nishita M, Tomizawa C, Yamamoto M, Horita Y, Ohashi K, Mizuno K. Spatial and temporal regulation of cofilin activity by lim kinase and slingshot is critical for directional cell migration. J Cell Biol. 2005;171(2):349–359.16230460 10.1083/jcb.200504029 PMC 2171197 · doi ↗ · pubmed ↗