Dystrophin protein and mRNA analyses for the molecular genetic diagnosis of dystrophinopathy: A novel deep intronic DMD variant
Zhiying Xie, Chang Liu, Qingyue Yuan, Zhihao Xie, Meng Yu, Yun Yuan

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
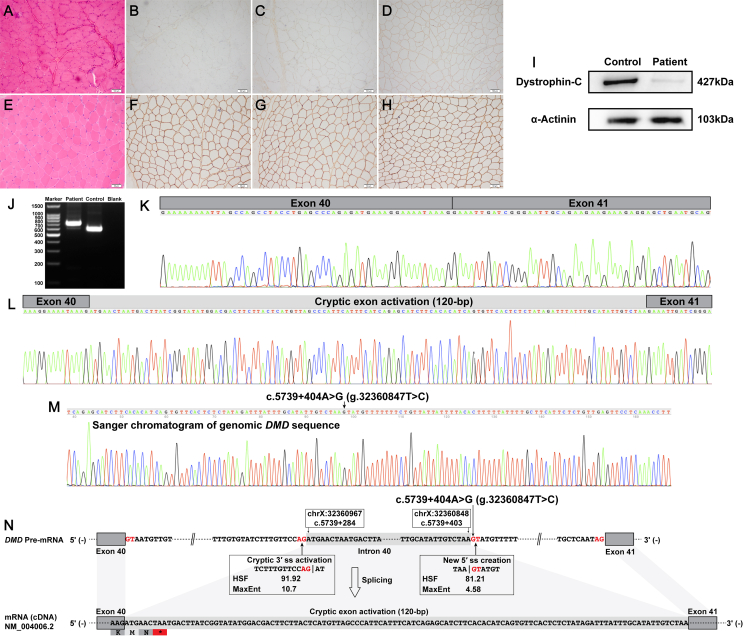 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMuscle Physiology and Disorders · Connective tissue disorders research · RNA modifications and cancer
Becker muscular dystrophy (BMD) and Duchenne muscular dystrophy (DMD) are X-linked recessive muscular dystrophies caused by pathogenic dystrophin (DMD) variants.1 The prevalence of DMD and BMD in Caucasian communities was estimated to be approximately 4∼6 per 100 000 people, which was similar to the prevalence in Asian communities.2 Deletions and/or duplications of one or more canonical exons account for ∼80% of disease-causing variants in DMD. Most of the remaining ∼20% pathogenic dystrophin variants are subexonic small variants, which include small insertions and/or deletions, missense variants, nonsense variants, and canonical splice site variants. Multiplex ligation-dependent probe amplification analysis combined with genomic sequencing of all DMD canonical exons and flanking intronic sequences (referred to as the routine DNA-based techniques) can identify most exonic deletions, exonic duplications, and subexonic small variants that occur in DMD canonical exons and/or adjacent intronic sequences.1 Some rare and atypical pathogenic dystrophin variants can escape the detection of routine DNA-based techniques, which mainly consist of complex chromosomal rearrangements and deep intronic splicing-altering variants.1 Cryptic exon-activating variants are the most common type among deep intronic splicing-altering variants in DMD.1 Here, a novel deep intronic cryptic exon-activating variant in the human dystrophin gene (NM_004006.2:c.5739 + 404A > G) was identified by skeletal muscle tissue-derived dystrophin protein and mRNA analyses, genomic Sanger sequencing, and long-read whole DMD gene sequencing.
A 30-year-old male patient, who had progressive muscle weakness of bilateral lower limbs since 25 years of age, was recruited. The level of his serum creatine kinase concentration was significantly increased (5878 IU/L). He now has bilateral tendon contractures, calf hypertrophy, and limb-girdle muscle weakness. However, he has no cardiac muscle involvement in terms of his clinical manifestations, electrocardiogram, and echocardiography examinations. Skeletal muscle biopsy revealed a mild muscular dystrophic pattern (Fig. 1A) and reduced expression of dystrophin-N, –C, and –R (Fig. 1B–D). Western blotting also revealed a reduced expression of full-length dystrophin protein in the recruited patient (6.7% of that found in the healthy control muscle; Fig. 1I). Multiplex ligation-dependent probe amplification analysis of dystrophin gene did not detect any deletions and/or duplications of one or more canonical exons in DMD. Whole-exome sequencing also did not detect any disease-causing genomic variants in him. Skeletal muscle tissue-derived dystrophin mRNA analysis revealed two different transcripts (Fig. 1J). TA cloning successfully distinguished the specific sequences in the two different transcripts; one was the normal splicing of DMD exons 40 to 41 (Fig. 1K) and the other was an abnormal 120-bp insertion sequence between DMD exons 40 and 41 (Fig. 1L). The abnormal 120-bp insertion sequence was homologous to (100%) a deep intronic region of DMD intron 40 (chrX:32360848–32360967) and thus was genetically regarded as a cryptic exon or pseudoexon activation (NM_004006.2:r.[=,5739_5740ins5739 + 284_5739 + 403]). The abnormal cryptic exon transcript, which encoded a frameshift and premature termination codon (NP_003997.1:p.[ = ,Glu1914Metfs∗3]), could be targeted for degradation by nonsense-mediated decay (Fig. 1N).Figure 1. Muscle pathologic findings, dystrophin mRNA studies, and genomic Sanger validation of the patient. (A) Hematoxylin-eosin staining showed a mild muscular dystrophic pattern. (B–D) Immunohistochemical staining showed no isolated positive myofibers and traces of dystrophin-N (B), a partial to severe reduction of dystrophin-C (C), and a mild to partial reduction of dystrophin-R (D). (E–H) Hematoxylin-eosin and immunohistochemical staining showed no histopathologic changes and positive and normal expression of dystrophin protein. (I) Western blotting revealed a reduced expression of full-length dystrophin protein. (J) Reverse transcription PCR amplification of the abnormal DMD splicing transcripts of our recruited patient showed two different bands. (K, L) TA cloning distinguished two different transcripts, including the wild-type DMD transcript (K) and an insertion sequence between DMD exons 40 and 41 (L). (M) Genomic Sanger validation of the novel deep intronic variant c.5739 + 404A > G in DMD. (N) Schematic diagram of the activated cryptic exon caused by c.5739 + 404A > G. (A–D) and (I) are for our recruited patient and (E–H) are for a healthy control. 3′ ss, acceptor splice site; 5′ ss, donor splice site; ∗, premature termination or stop codon; MaxEnt, maximum entropy; HSF, human splicing finder.Figure 1
Long-read whole DMD gene sequencing1 of the patient identified a novel and de novo single-nucleotide variation around the activated DMD cryptic exon (Fig. S1), i.e., the NM_004006.2:c.5739 + 404A > G variant, which was further validated by genomic Sanger sequencing (Fig. 1M). In addition, no pathogenic structural DMD variants were identified by the long-read sequencing. The novel c.5739 + 404A > G variant created a new deep intronic 5′ ss (donor splice site) (TAA|GTATGT) and activated a pre-existing cryptic 3′ ss (acceptor splice site; TCTTTGTTCCAG|AT) in intron 40 of the dystrophin gene (Fig. 1N). The SpliceAI algorithm predicted the same splicing alterations caused by c.5739 + 404A > G as the Human Splicing Finder and Maximum Entropy algorithms: Donor Gain for the new donor splice site (1-bp) and Acceptor Gain for the pre-existing cryptic acceptor splice site (120-bp) in DMD intron 40. The activation of a pre-existing cryptic 3′ ss and the creation of a new deep intronic 5′ ss in DMD intron 40 eventually caused the activation of a new DMD cryptic exon identified in our recruited patient (Fig. 1N). The details of skeletal muscle biopsy, western blotting, dystrophin mRNA analysis, TA cloning, long-read whole DMD gene sequencing, and bioinformatic analyses are described in supplementary information.
Pathogenic variants located in introns of protein-coding genes are frequently found to alter pre-mRNA splicing and then cause abnormalities in mRNA and protein. Pathogenic splicing-altering variants account for approximately 9% of all identified pathogenic variants associated with monogenic diseases, including exonic and intronic variants with splicing impact.3 Most intronic splicing-altering variants are located in adjacent exon-intron boundaries; hence, they can weaken the strength of essential splicing signals of canonical exons and cause various abnormal pre-mRNA splicing events.4 In addition, some intronic splicing-altering variants can also occur in the deep intronic regions of protein-coding genes, and in this condition, they may cause the inclusion of deep intronic sequences into the mature transcripts, which refer to the activation of cryptic exons or pseudoexons. The mechanisms underlying cryptic exon activation mainly include the alterations in splicing regulatory elements and/or essential splicing signals within deep intronic regions.4 Deep intronic splicing-altering variants are increasingly reported to cause cryptic exon activation in protein-coding genes associated with monogenic diseases, such as ABCA4, COL4A5, DMD, CCN6, DYSF, and COL6A1.4 Among them, the DMD gene has a higher occurrence rate of deep intronic splicing-altering variants compared with other monogenic genes, as the DMD intronic sequences with an extremely large genomic size provides a bigger target for deep intronic mutations.4 To our knowledge, deep intronic splicing-altering variants account for about 3% of all pathogenic DMD variants associated with BMD and DMD.1 In our study, we found a novel deep intronic splicing-altering variant, the c.5739 + 404A > G variant in DMD intron 40, which activated a new dystrophin cryptic exon and confirmed the genetic diagnosis of BMD in our recruited patient.
Our study further indicates that muscle biopsy is still an important diagnostic approach for patients with a suspected muscle disease and without a definite genetic diagnosis in the genomic era. Muscle-derived protein and mRNA studies allow the detection and interpretation of nearly all types of pathogenic variants associated with inherited muscle diseases, including atypical missense and synonymous variants, structural variants, non-canonical splice site variants, and deep intronic variants.1 Muscle-derived mRNA analysis can also confirm the impact of genomic DNA variants on pre-mRNA splicing to identify abnormalities in splice sites, branch points, or splicing regulatory elements.4 In addition, it allows the interpretation of the open reading frame conservation that determines the clinical severity of monogenic diseases. In addition to the main determinant factor (the open reading frame conservation), other factors including epigenetic changes and residual levels of wild-type transcripts should also be considered when interpreting patients' clinical severity. As in our recruited patient, the novel deep intronic DMD variant produced an out-of-frame transcript that could theoretically cause a DMD phenotype; however, the residual level of normally spliced DMD transcript detected in the patient alleviated his clinical severity and resulted in his BMD phenotype. As skin biopsies are less invasive than muscle biopsies, skin biopsy-derived fibroblasts are a good investigation source for detecting aberrant DMD splicing transcripts after converting to myogenic cells,5 which might be an alternative tool for mRNA-based genetic diagnosis of BMD and DMD.
Our case study identified a novel deep intronic DMD variant via the stepwise application of whole-exome sequencing, dystrophin protein and mRNA analyses, genomic Sanger sequencing, and long-read sequencing, highlighting the significance of intronic DMD variants in genetically undiagnosed BMD patients.
CRediT authorship contribution statement
Zhiying Xie: Writing – review & editing, Writing – original draft, Visualization, Validation, Methodology, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization. Chang Liu: Writing – review & editing, Writing – original draft, Visualization, Validation, Methodology, Investigation, Formal analysis, Data curation. Qingyue Yuan: Writing – review & editing, Software, Methodology, Investigation. Zhihao Xie: Writing – review & editing, Validation, Software, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Meng Yu: Writing – review & editing, Validation, Supervision, Software, Methodology, Investigation, Data curation, Conceptualization. Yun Yuan: Writing – review & editing, Visualization, Validation, Supervision, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization.
Ethics declaration
This study was approved by the Ethics Committee at Peking University First Hospital (approval number: 2023 109–002). The patient provided informed consent for the publication of this case.
Funding
This work was supported by the 10.13039/501100001809National Natural Science Foundation of China (No. 82201553) and the National High Level Hospital Clinical Research Funding (China) (High Quality Clinical Research Project of 10.13039/100017415Peking University First Hospital, No. 2023HQ10; Scientific Research Fund of 10.13039/100017415Peking University First Hospital).
Conflict of interests
The authors declared no competing interests.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Xie Z.Sun C.Liu C.Clinical, muscle imaging, and genetic characteristics of dystrophinopathies with deep-intronic DMD variants J Neurol 270220239259373631976810.1007/s 00415-022-11432-0 · doi ↗ · pubmed ↗
- 2Salari N.Fatahi B.Valipour E.Global prevalence of Duchenne and Becker muscular dystrophy: a systematic review and meta-analysis J Orthop Surg Res 1712022963516864110.1186/s 13018-022-02996-8PMC 8848641 · doi ↗ · pubmed ↗
- 3Stenson P.D.Mort M.Ball E.V.The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies Hum Genet 136620176656772834924010.1007/s 00439-017-1779-6PMC 5429360 · doi ↗ · pubmed ↗
- 4Keegan N.P.Wilton S.D.Fletcher S.Analysis of pathogenic pseudoexons reveals novel mechanisms driving cryptic splicing Front Genet 12202280694610.3389/fgene.2021.806946 PMC 881918835140743 · doi ↗ · pubmed ↗
- 5Rossi R.Torelli S.Ala P.Myo D-induced reprogramming of human fibroblasts and urinary stem cells in vitro: protocols and their applications Front Physiol 142023114504710.3389/fphys.2023.1145047 PMC 1022978337265839 · doi ↗ · pubmed ↗