Synthesis of a Functionalized Bicyclo[3.2.1]Octane: A Common Subunit to Kauranes, Grayananes, and Gibberellanes
Nicolas Fay, Camil Benbouziyane, Cyrille Kouklovsky, Aurélien de la Torre

TL;DR
This paper describes a successful method to synthesize a shared building block found in three types of natural diterpenoids.
Contribution
A novel synthetic route using ring-closing metathesis to build a bicyclo[3.2.1]octane scaffold common to kauranes, grayananes, and gibberellanes.
Findings
An initial 1,4-sila-Prins cyclization approach failed, yielding an undesired 1,2-cyclization product.
A successful synthesis was achieved in 8 steps using ring-closing metathesis (RCM).
The resulting scaffold is a functionalized bicyclo[3.2.1]octane suitable for further natural product synthesis.
Abstract
Kauranes, grayananes, and gibberellanes are three important diterpenoid families. These natural products all share a bicyclo[3.2.1]octane skeleton, with oxidation at very specific positions. In this manuscript, we describe the synthesis of a bicyclo[3.2.1]octane building block, which could serve as a potential intermediate for the synthesis of natural products from these three families. A first approach relying on a 1,4‐sila‐Prins cyclization was first explored, which required the selective functionalization of dihydrocarvone (via C─H activation and selective oxidation). This strategy resulted in a dead end when a 1,2‐cyclization product was obtained. An alternative strategy relying on ring‐closing metathesis (RCM) allowed to successfully achieve the synthesis of the desired scaffold in 8 steps from cyclohexenone. A detailed account on the synthesis of a bicyclo[3.2.1]octane scaffold,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Scheme 1
Scheme 1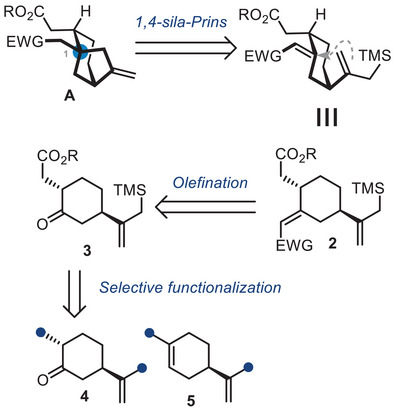 Scheme 2
Scheme 2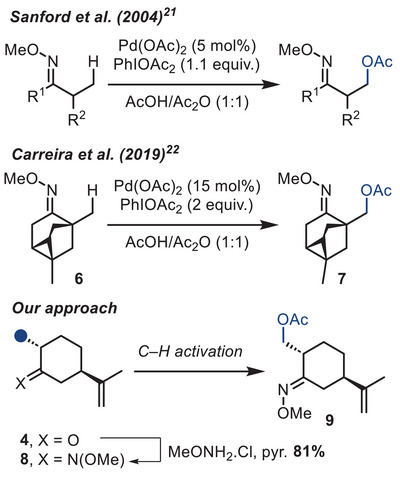 Scheme 3
Scheme 3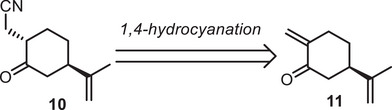 Scheme 4
Scheme 4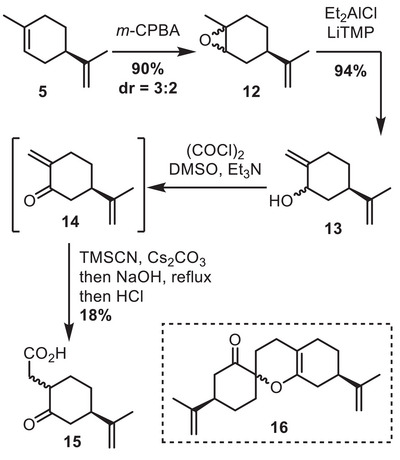 Scheme 5
Scheme 5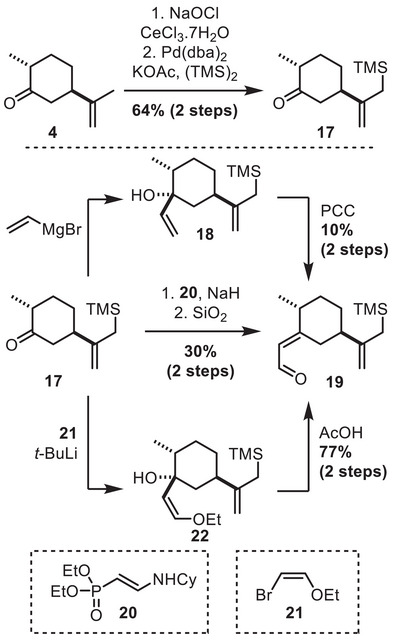 Scheme 6
Scheme 6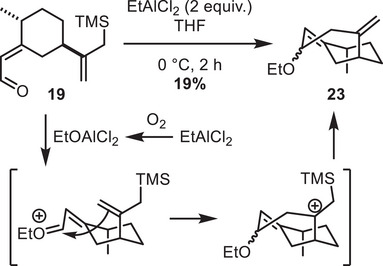 Scheme 7
Scheme 7 Scheme 8
Scheme 8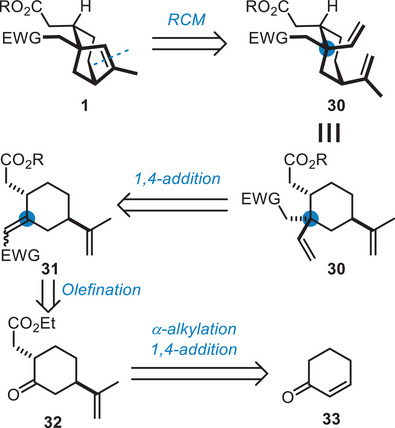 Scheme 9
Scheme 9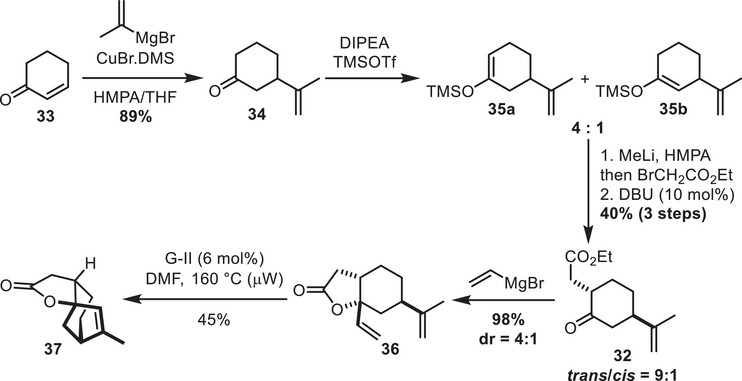 Scheme 10
Scheme 10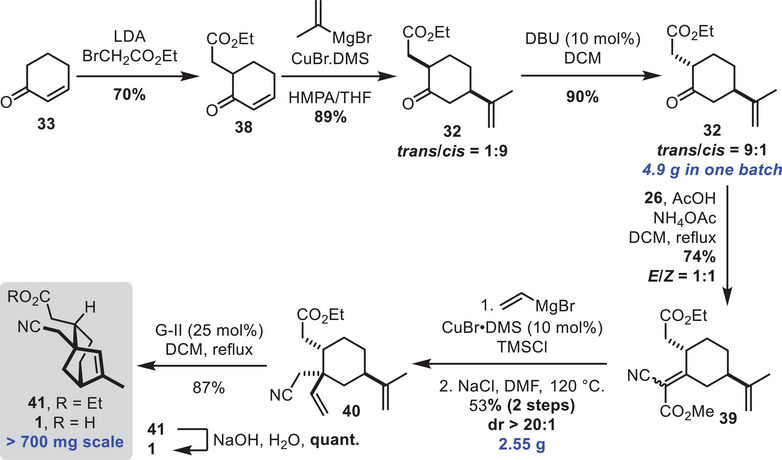 Scheme 11
Scheme 11|
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | [Pd] [x mol%] | y | Solvent | Temperature | Time | Conversion [%] | Yield [%][
|
| 1 | Pd(OAc)2 (5 mol%) | 1.1 | AcOH/Ac2O (1:1) | 70 °C | 3 hours | 20 | 10 |
| 2 | Pd(OAc)2 (5 mol%) | 1.1 | AcOH/Ac2O (1:1) | 70 °C | 16 hours | 80 | 18 |
| 3 | Pd(OAc)2 (5 mol%) | 1.1 | AcOH/Ac2O (1:1) | 100 °C | 3 hours | 100 | ND |
| 4 | Pd(OAc)2 (5 mol%) | 1.1 | AcOH/Ac2O (1:1) | 100 °C | 1.5 hours | 100 | 21 |
| 5 | Pd(OAc)2 (5 mol%) | 1.1 | AcOH/Ac2O (1:1) | 100 °C | 1 hour | 100 | 26 |
| 6 | Pd(OAc)2 (20 mol%) | 1.1 | AcOH/Ac2O (1:1) | 100 °C | 1 hour | 100 | 22 |
| 7 | Pd(OAc)2 (5 mol%) | 2 | AcOH/Ac2O (1:1) | 100 °C | 1 hours | 100 | ND |
| 8 | Pd(OAc)2 (5 mol%) | 1.1 | AcOH/Ac2O (1:1) | 100 °C | 15 minutes | 100 | ND |
| 9 | Pd(dba)2 (5 mol%) | 1.1 | AcOH/Ac2O (1:1) | 100 °C | 1 hour | 100 | 32 |
| 10 | Pd(dba)2 (5 mol%) | 1.1 | MeCN | 100 °C | 1 hour | 100 | ND |
- —Agence Nationale de la Recherche10.13039/501100001665
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBioactive Natural Diterpenoids Research · Synthetic Organic Chemistry Methods · Marine Sponges and Natural Products
Introduction
1
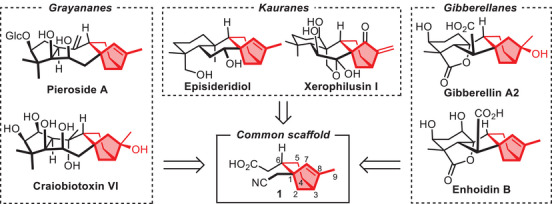
Kauranes are a broad family of diterpenoids encountered in the Lamiaceae, Asteraceae, Annonaceae, and Euphorbiaceae families.^[^ 1, 2 ^]^ These natural products display a wide range of biological activities, including antibacterial, anticancer, and antifungal, providing them with therapeutic potential. Kauranes have a tetracyclic structure with 6/6/6/5‐membered rings, and various possible oxidation patterns (Scheme 1). Their synthesis has attracted the attention of the organic chemistry community, with reports as early as 1962 by Ireland et al,^[^ 3 ^]^ and a continuous interest since then.^[^ 4, 5, 6 ^]^ Grayananes are a biosynthetically related family of natural products, originating from a^[^ 1, 2 ^]^‐rearrangement from the kaurane skeleton, which leads to a tetracycle with 5/7/6/5‐membered rings.^[^ 7 ^]^ Grayananes are mainly found in the Ericaceae plant family, which are commonly used in traditional medicine around the world. They display interesting antinociceptive, antifeedant, and antitumor activities, which prompted various groups to engage in their total synthesis,^[^ 8 ^]^ from early relay synthetic approaches by Matsumoto^[^ 9 ^]^ to recent strategies by Newhouse,^[^ 10 ^]^ Ding,^[^ 11 ^]^ Luo,^[^ 12 ^]^ Jia,^[^ 13, 14 ^]^ and Yang.^[^ 15 ^]^ The gibberellane family is another biosynthetically related diterpenoid family, originating from a different^[^ 1, 2 ^]^‐rearrangement from a functionalized kaurane precursor.^[^ 16 ^]^ They are produced by various plants, bacteria, and fungal species. The first synthesis of gibberellanes was reported by Mori at the same time as the early kaurane syntheses,^[^ 17 ^]^ but some gibberellanes were also recently synthesized by Dai and Fan.^[^ 18, 19 ^]^ These three natural product families display a broad diversity of structures owing to the multiple possible oxidation patterns.
Structure of kauranes, grayananes, gibberellanes, and proposed common synthetic scaffold.
Natural products from these three families share a bicyclo[3.2.1]octane scaffold, often named rings C and D. The synthesis of this scaffold represents a challenge which has stimulated the creativity of organic chemists for decades.^[^ 20 ^]^ Attracted by the prospect of reaching various natural products within these three different natural product families, we decided to design a common intermediate 1 having the shared bicyclo[3.2.1]octane scaffold. In order to introduce potential oxidation at C^7^, C^8^, or C^9^, we imagined an olefin at C^7^═C^9^, which could undergo Mukaiyama hydration^[^ 21 ^]^ or epoxidation/elimination.^[^ 22 ^]^ Moreover, we wanted to have two orthogonal chemical handles, which would allow to build the rest of the scaffolds. Thus, we designed this intermediate with a carboxylic acid and a nitrile group, which can be functionalized chemoselectively. In this manuscript, we describe our efforts toward the synthesis of this scaffold.
Results and Discussion
2
The key challenge is the formation of the bicyclo[3.2.1]octane scaffold containing the quaternary center C^1^ at the bridgehead position. Our original approach relied on a 1,4‐sila‐Prins cyclization reaction for the formation of A, which would have an exocyclic olefin allowing possible oxidation,^[^ 21 ^]^ from intermediate 2 having the cyclohexane core, an allylsilane, and a Michael acceptor (Scheme 2). We imagined that the Michael acceptor could be obtained from the corresponding ketone 3, which we could trace back to either dihydrocarvone 4 or limonene 5.
Proposed retrosynthesis of 1.
C─H Activation from Dihydrocarvone
2.1
In our initial approach, we consider valorizing natural dihydrocarvone 4, readily available from commercial sources in enantioenriched form. The C─H activation method developed by Sanford in 2004, allowing the β‐functionalization of oximes (Scheme 3), seemed particularly suited for that.^[^ 23 ^]^ In particular, Carreira and coworkers had also applied this method to the functionalization of more complex 2‐methylcyclohexanone derivative 6 in the synthesis of the tricyclooctane core of trachylobane natural products.^[^ 24 ^]^ To test this approach, we prepared the O‐methyloxime derivative 8 from dihydrocarvone 4, and submitted it to various C─H activation conditions.
Oxime directed C─H acetoxylation.
Our first attempts were directly based on the reaction conditions described by Sanford et al., using Pd(OAc)2 as the catalyst and PhI(OAc)2 as the oxidant in a mixture of AcOH/Ac_2_O (Table 1). The reaction required heating at 100 °C to reach full conversion, although prolonged reaction times were detrimental to the reaction (entries 1–5). Increasing catalyst loading (entry 6) did not significantly improve the yield, while increasing the amount of oxidant led to over‐oxidation products (entry 7). The best result was obtained using Pd(dba)2 as the palladium source (entry 9), while heating with microwave irradiation or changing the solvent to acetonitrile led to decomposition (entries 8 and 10).
In all cases, although the conversion was complete, product 9 was obtained in low yield. We assume that this is due to the presence of an olefin as well as inherently more reactive allylic C─H, although no other product could be isolated. It should be noted that neither Sanford nor Carreira had tested this reaction on substrates bearing olefins. This limitation led us to abandon the C─H activation approach.
Hydrocyanation Reaction from a Limonene Derivative
2.2
As an alternative approach, we imagined a key 1,4‐hydrocyanation reaction to introduce a nitrile moiety on an enone derivative 11 (Scheme 4). The enone could itself be traced back to commercially available limonene 5, also accessible in enantioenriched form.
Proposed 1,4‐hydrocyanation.
To test this approach, we converted limonene 5 into perillyl alcohol 13 through an epoxidation/elimination sequence developed by Evans.^[^ 25 ^]^ Perillyl alcohol was then oxidized under Swern conditions. The resulting enone 14 was very unstable and had to be engaged directly into the key 1,4‐hydrocyanation. As the hydrocyanation product was unstable and the reaction was reversible, we had to further hydrolyze the nitrile into the corresponding carboxylic acid 15. Despite all our attempts, the product was obtained at best with 18% yield. This was due to the formation of side product 16 arising from a hetero‐Diels‐Alder dimerization, which had previously been described by Hayes in the synthesis of cymbodiacetal.^[^ 26 ^]^
The poor yields obtained in this reaction led us to abandon this approach, and explore alternative strategies not relying on the chiral pool (Scheme 5).
Attempts toward the 1,4‐hydrocyanation approach.
Efforts toward the Formation of the Bicyclo[3.2.1]Octane
2.3
Our strategy relied on a key 1,4‐Sakurai reaction for the bicyclo[3.2.1]octane construction. Although challenging due to the poor orbital alignment inherent to this 5‐enolendo‐exo‐trig cyclization mode,^[^ 27 ^]^ some precedents in the literature indicated that a similar cyclization could proceed with the right Lewis acid.^[^ 28, 29 ^]^ To test this approach, a model substrate for this cyclization was elaborated from dihydrocarvone 4. Allylic chlorination followed by palladium‐catalyzed silylation allowed to introduce the allylsilane moiety (Scheme 6). Following this, ketone 17 underwent a vinyl Grignard addition, followed by chromium‐mediated Babler‐Dauben oxidative rearrangement. However, this sequence led to poor yield due to undesired protodesilylation (see Supporting Information for detail). As an alternative, we tested Nagata homologation using reagent 20. Again, the product 19 was obtained in unsatisfying yield. Finally, addition of the organolithium reagent deriving from 21 followed by acid hydrolysis provided 19 in good yield. A careful choice of acidic conditions was necessary in order to minimize the competitive protodesilylation in this process (see Supporting Information for more detail).
Preparation of a model substrate for the 1,4‐sila‐Prins reaction.
With compound 19 in hand, we started to explore the key 1,4‐sila‐Prins cyclization. While most conditions afforded protodesilylation or degradation of 19 (see the Supporting Information for more detail), the use of EtAlCl_2_ in THF led to the unexpected formation of bicyclo[4.3.1]decane 23 (Scheme 7). We rationalize this by the presence of trace amounts of oxygen in THF, which could oxidize EtAlCl_2_ into EtOAlCl_2_, promoting the formation of an oxonium, which would then undergo a 1,2‐sila‐Prins cyclization. This observation indicates that 1,2‐sila‐Prins is geometrically favored over 1,4‐addition on this scaffold, and prompted us to explore other functionalities to achieve the desired cyclization.
Unexpected 1,2‐sila‐Prins reaction.
In order to explore other substrates for cyclization in which formation of the bicyclo[4.3.1]decane would not be a competitive pathway, we synthesized Michael acceptors 25 and 27 by Horner‐Wadsworth‐Emmons and Knoevenagel reactions, respectively, from 17 (Scheme 8). Moreover, epoxide 28 was prepared by a Corey‐Chaykovsky reaction, and propargyl alcohol 29 was obtained by addition of lithiated TMS‐acetylene. All these products were obtained as diastereomeric mixtures. However, all attempts to cyclize these alternative substrates led either to decomposition or protodesilylation (see Supporting Information for more details).
Preparation of alternative cyclization substrates.
The challenges associated with this cyclization can be explained by the cyclization mode, which is close to a 5‐enolendo‐exo‐trig, unfavored according to Baldwin's rules modification relative to enolates.^[^ 27 ^]^ This can be rationalized by a poor orbital overlap between the two π‐systems due to geometrical constrains.^[^ 30, 31 ^]^ After reaching these multiple dead ends, we decided to turn away from the cyclization approach and explore a different strategy based on ring‐closing metathesis (RCM).
Synthesis of the Bicyclo[3.2.1]Octane Framework by Ring‐closing Metathesis
2.4
A revised strategy relying on RCM was designed, in which 1 would be obtained from 30 having the two olefins appended for the key RCM (Scheme 9). The quaternary stereogenic center would be formed by a diastereoselective 1,4‐addition from Michael acceptor 31, which would in turn be formed from functionalized cyclohexanone 32. For a fast preparation of the functionalized cyclohexanone intermediate 32, we considered a two‐step approach from cyclohexenone 33 involving α‐alkylation of the ketone and 1,4‐addition. Both orders of steps can be envisaged: achieving the α‐alkylation first could simplify the regioselectivity question in the enolate formation, while achieving the 1,4‐addition first would allow an enantioselective synthesis using the conditions developed by Schmalz.^[^ 32 ^]^
Revised strategy relying on RCM.
Cyclohexenone 33 underwent a Cu‐catalyzed 1,4‐addition to introduce the isopropenyl function (Scheme 10). It should be noted that the enantioselective version of this reaction was reported by Schmalz with very high levels of enantioselectivity.^[^ 32 ^]^ As demonstrated by Vanderwal and coworkers,^[^ 33 ^]^ soft enolization could allow the formation of the enol ether 35 with a decent regioselectivity (for more details on the enolization conditions explored, see the Supporting Information). Treatment of this enol ether with MeLi and trapping the resulting lithium enolate with ethyl‐α‐bromoacetate followed by DBU‐mediated epimerization ultimately led to the desired functionalized cyclohexanone 32. To further test the viability of the RCM approach, we subjected ketone 32 to vinyl Grignard addition. The resulting lactone 36 could cyclize in the presence of Grubbs 2^nd^ generation catalyst under microwave conditions, leading to the tricyclic structure 37.
Functionalization of cyclohexanone by performing first the conjugate addition.
A faster way to prepare 32 was achieved by reversing the step order. Starting from cyclohexenone 33, α‐alkylation with ethyl‐α‐bromoacetate led to the formation of 38, which further underwent a Cu‐catalyzed 1,4‐addition to introduce the isopropylidene moiety (Scheme 11). This led to product 32 with undesired cis‐diastereoselectivity. The desired trans arrangement could be obtained by simply treating the product with DBU to epimerize the stereogenic center α to the ketone. More importantly, this sequence could be easily scaled‐up to multigram scale. Then, the ketone precursor 32 underwent a Knoevenagel condensation. The resulting α,β‐unsaturated cyanoester 39 was subjected to a Cu‐catalyzed 1,4 addition using a vinyl Grignard reagent. To simplify the diasteromeric mixture, the methyl ester was decarboxylated under Krapcho conditions (see Supporting Information for details). The polysubstituted cyclohexane 40 having the required vinyl moieties for a ring closing metathesis was obtained with excellent diastereoselectivity. Following that, the RCM went smoothly and the desired bicyclo[3.2.1]octane 41 was isolated in good yield. The ester moiety could further be hydrolyzed without affecting the nitrile, thus leading to the target fragment 1. Moreover, this synthesis could be easily scaled up to 700 mg.
Formation of the bicyclo[3.2.1]octane by ring‐closing‐metathesis.
Conclusion
3
To conclude, we have reported the synthesis of a functionalized bicyclo[3.2.1]octane scaffold, a skeleton common to various natural products from the kaurane, grayanane, and gibberellane families. This compound possesses two orthogonal functionalities (nitrile and carboxylic acid), which could be used for further elaboration of the above‐mentioned structures. Our first approaches relying on the chiral pool (starting from dihydrocarvone or limonene) proved unsuccessful. Moreover, the original plan of forming the bicyclo[3.2.1]octane by a 1,4‐sila‐Prins cyclization also failed, due to geometrical constrains, which could not be overcome. Ultimately, we successfully prepared the bicyclo[3.2.1]octane framework by a RCM strategy. The application of this building block to the total synthesis of natural products from the three aforementioned diterpenoid families will be studied in the future in our lab.
Supporting Information
The authors have cited additional references within the Supporting Information.^[^ 34, 35, 36, 37, 38, 39, 40, 41 ^]^
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1T. S. Ibrahim , P. Khongorzul , M. Muyaba , R. N. Alolga , Front. Pharmacol. 2023, 14, 1227574 and references therein.37456746 10.3389/fphar.2023.1227574 PMC 10345206 · doi ↗ · pubmed ↗
- 2S. Kibet , N. M. Kimani , S. S. Mwanza , C. M. Mudalungu , C. B. R. Santos , C. M. Tanga , Pharmaceuticals 2024, 17, 510 and references therein.38675469 10.3390/ph 17040510 PMC 11054903 · doi ↗ · pubmed ↗
- 3R. A. Bell , R. E. Ireland , R. A. Partyka ,J. Org. Chem. 1962, 27, 3741.
- 4X. Zhao , B. Cacherat , Q. Hua , D. Ma , Nat. Prod. Rep. 2022, 39, 119 and references therein.34263890 10.1039/d 1np 00028 d · doi ↗ · pubmed ↗
- 5G. Yue , B. Liu , Chem Plus Chem 2024, 89, e 202300676 and references therein.38414152 10.1002/cplu.202300676 · doi ↗ · pubmed ↗
- 6J. Guo , B. Li , W. Ma , M. Pitchakuntla , Y. Jia , Angew. Chem. Int. Ed. 2020, 59, 15195.10.1002/anie.20200593232427394 · doi ↗ · pubmed ↗
- 7C.‐H. Li , J.‐Y. Zhang , X.‐Y. Zhang , S.‐H. Li , J.‐M. Gao , Eur. J. Med. Chem. 2019, 166, 400 and references therein.30739823 10.1016/j.ejmech.2019.01.079 · doi ↗ · pubmed ↗
- 8N. Fay , R. Blieck , C. Kouklovsky , A. de la Torre , Beilstein J. Org. Chem. 2022, 18, 1707 and references therein.36570567 10.3762/bjoc.18.181PMC 9764858 · doi ↗ · pubmed ↗