Phage-encoded TelN inhibits bacterial Mre11-Rad50 nuclease to protect hairpin telomeres
Maya Houmel, Nicolas Pellaton, Anna Anchimiuk, Stephan Gruber

TL;DR
This paper shows how a phage protein called TelN protects its DNA ends from bacterial repair enzymes, revealing a new mechanism for telomere protection.
Contribution
The study identifies TelN as a key protector of hairpin telomeres by inhibiting Mre11-Rad50 nuclease activity in bacteria.
Findings
TelN is essential and sufficient to protect phage hairpin telomeres from Mre11-Rad50 processing.
Protection requires TelN's DNA binding and protein-protein interactions with the MR complex.
TelN's protection mechanism is independent of its resolution activity and C-terminal domains.
Abstract
Ends of linear chromosomes require protection from host repair machinery that otherwise will mistake them for damaged DNA. The E. coli bacteriophage N15 harbors a linear genome with covalently closed hairpin ends formed by the phage-encoded telomere resolvase TelN. The double-strand break repair complex Mre11-Rad50 (MR, SbcCD in E. coli) specifically targets DNA termini, yet how hairpin telomeres evade host nuclease degradation in bacteria remains unknown. Here, we demonstrate that TelN is essential and sufficient to protect N15 phage-derived hairpin telomeres from MR processing in E. coli. Using a combination of genetic and biochemical approaches, we show that this protective function requires both TelN sequence-specific DNA binding and species-specific protein-protein interactions. Notably, we found that protection is independent of TelN’s resolution activity and does not require the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
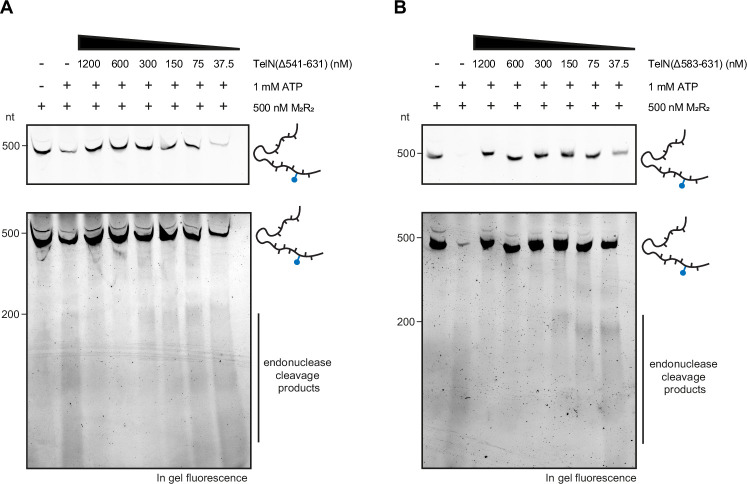 Figure 10
Figure 10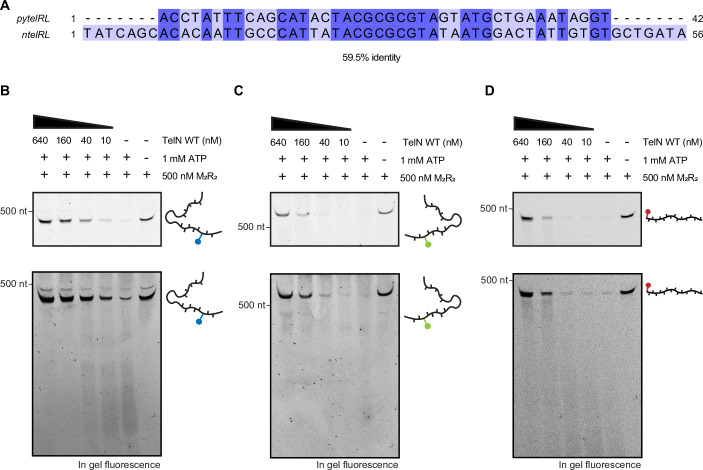 Figure 11
Figure 11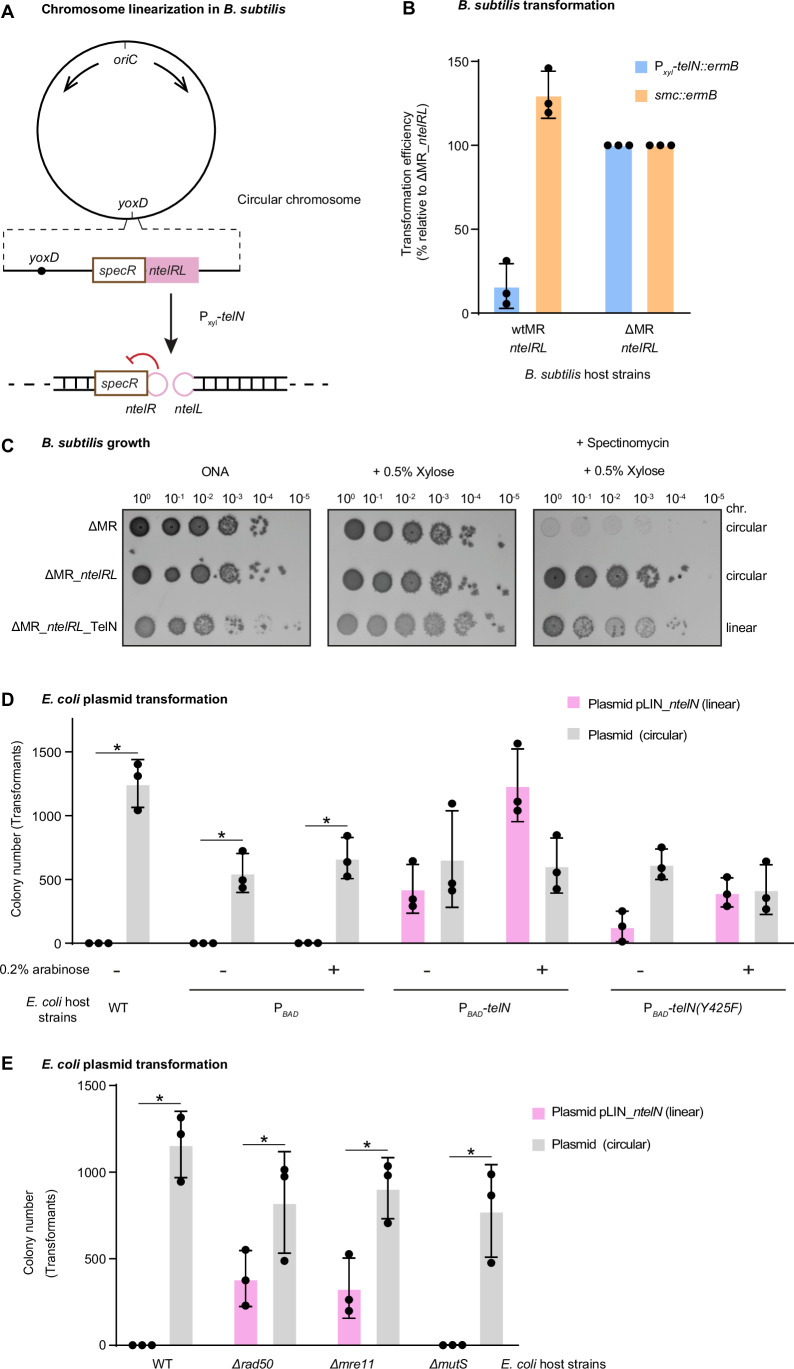 Figure 1
Figure 1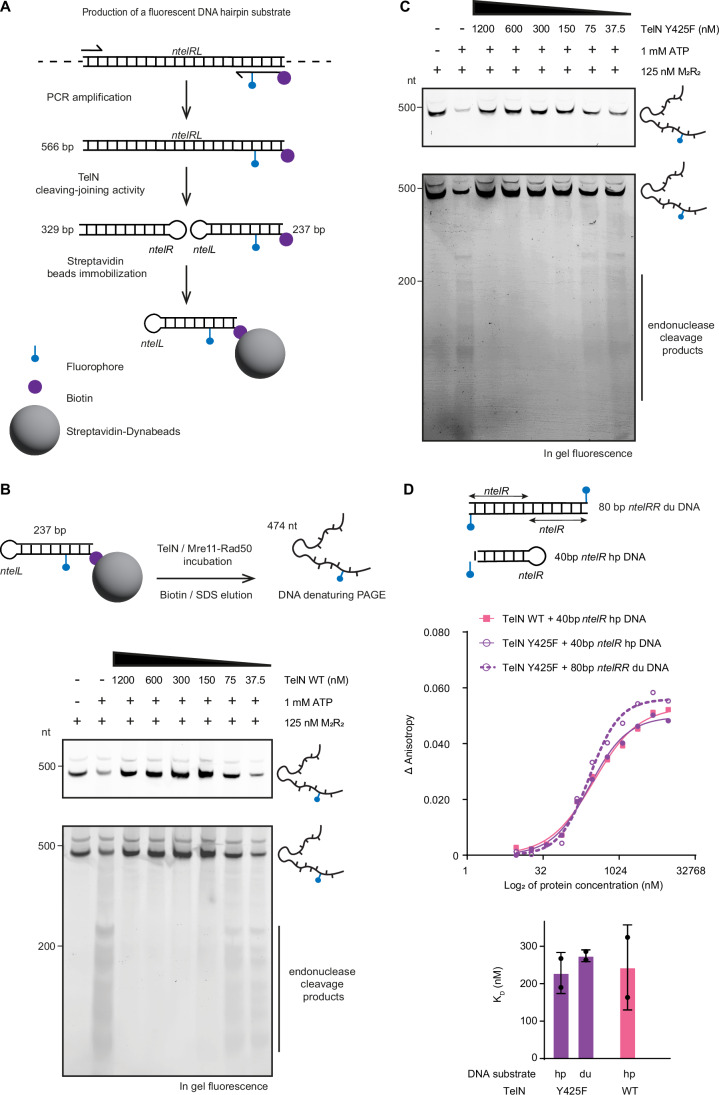 Figure 2
Figure 2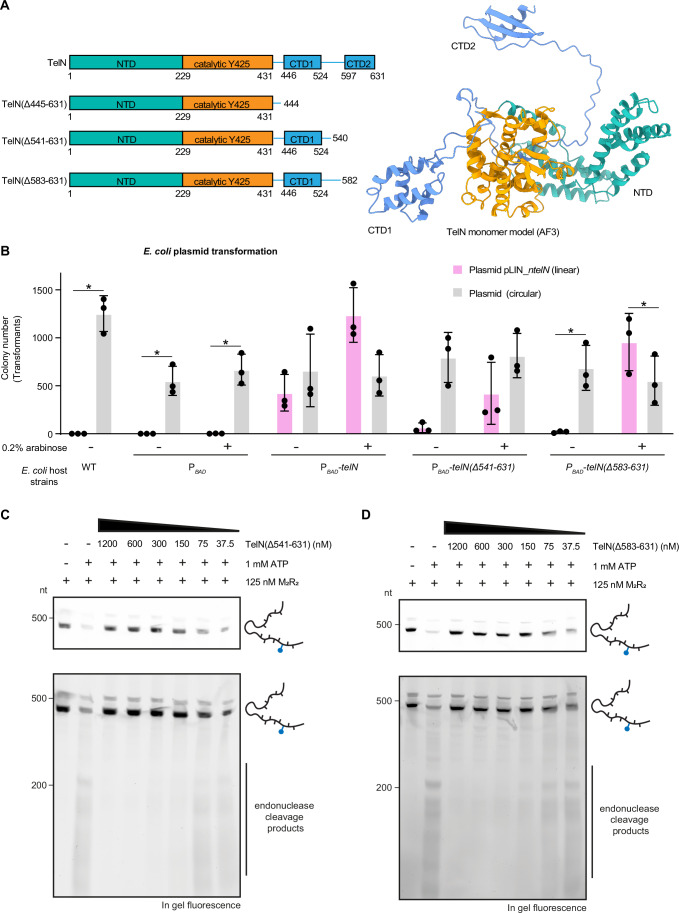 Figure 3
Figure 3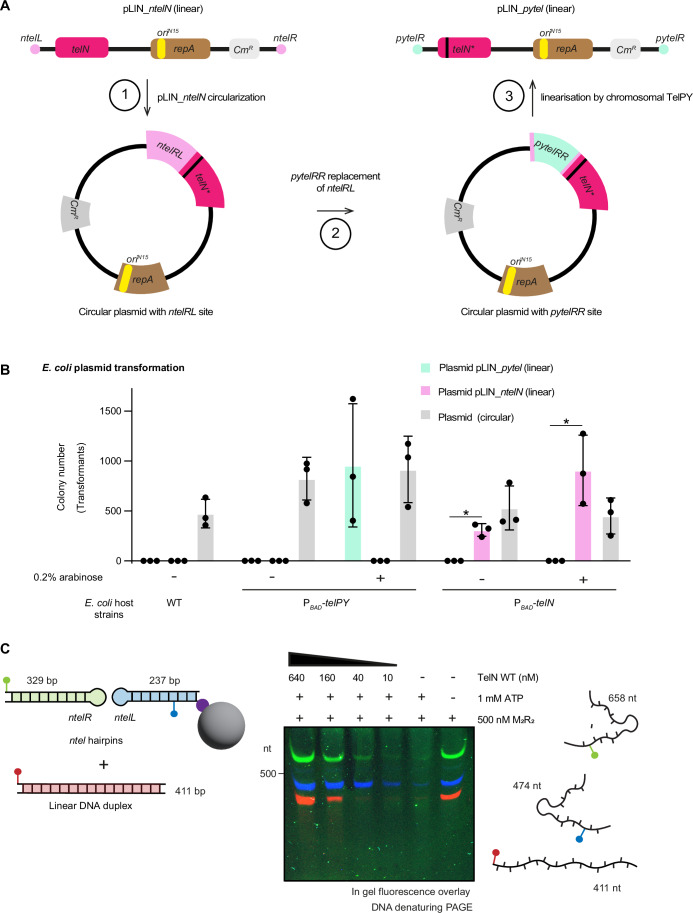 Figure 4
Figure 4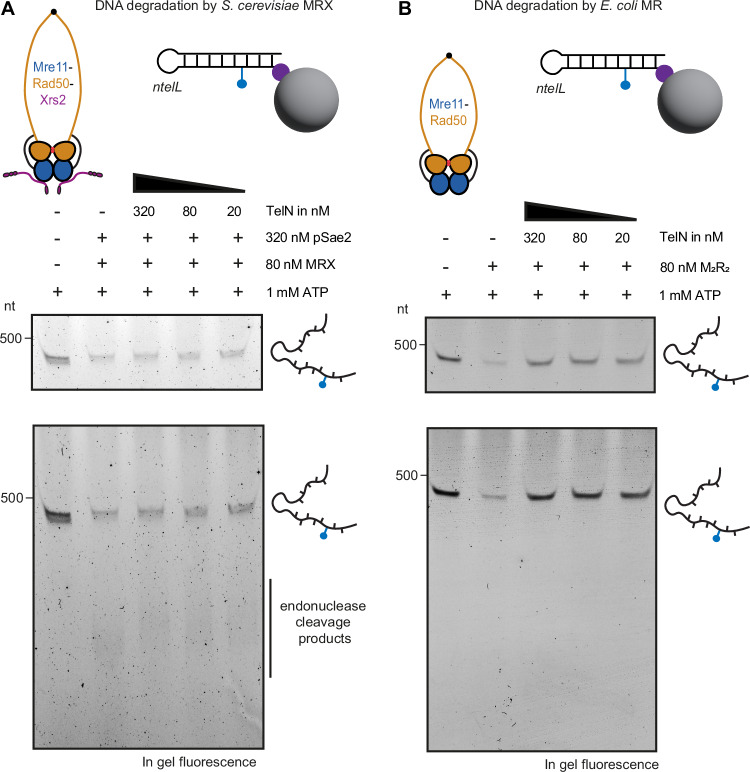 Figure 5
Figure 5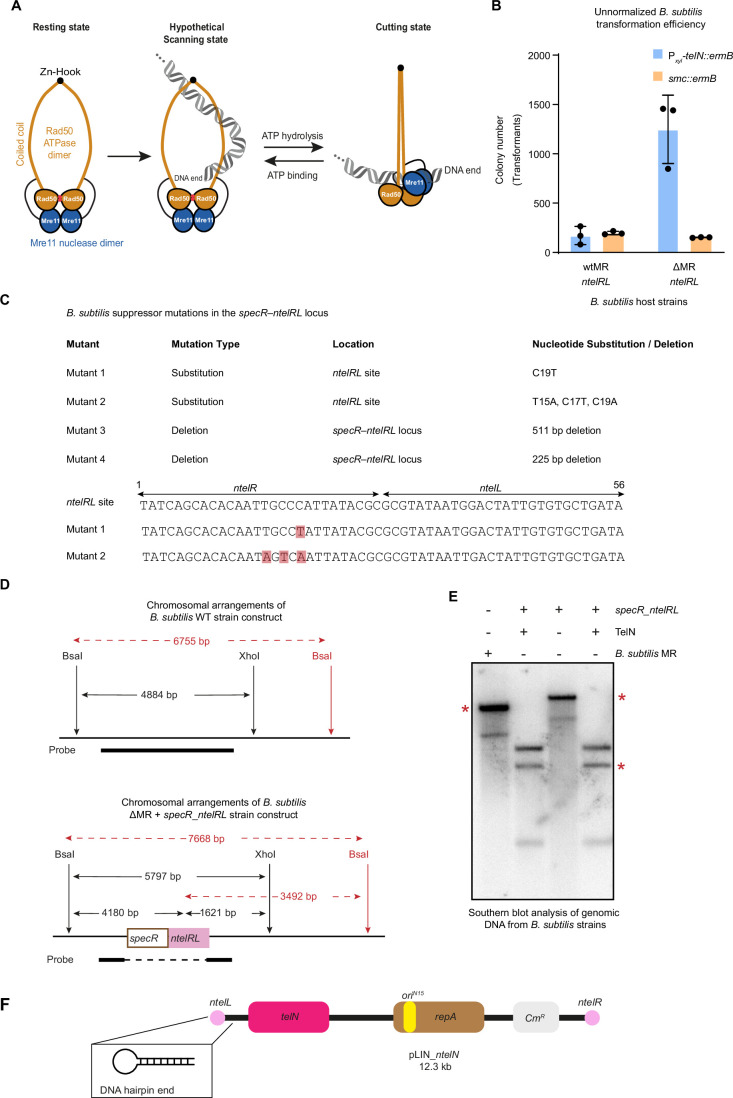 Figure 6
Figure 6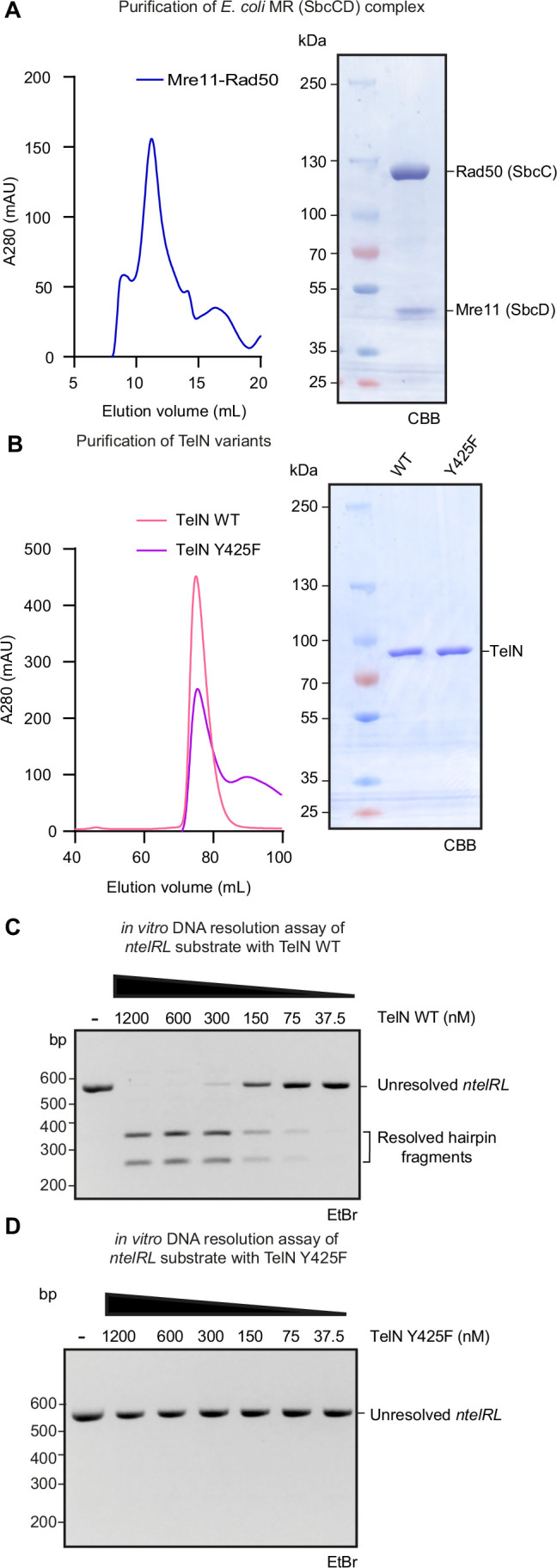 Figure 7
Figure 7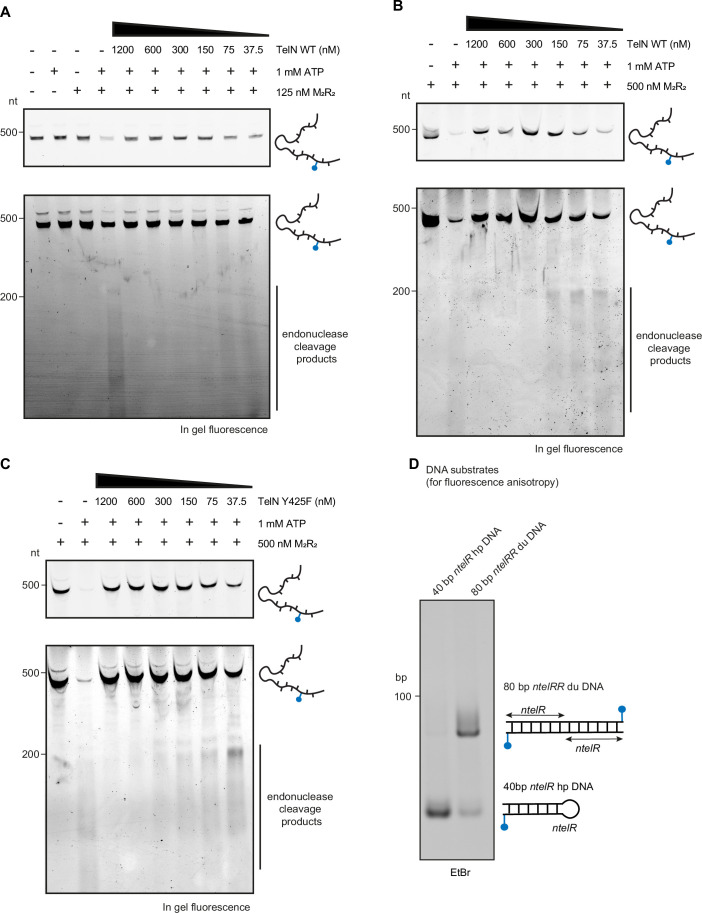 Figure 8
Figure 8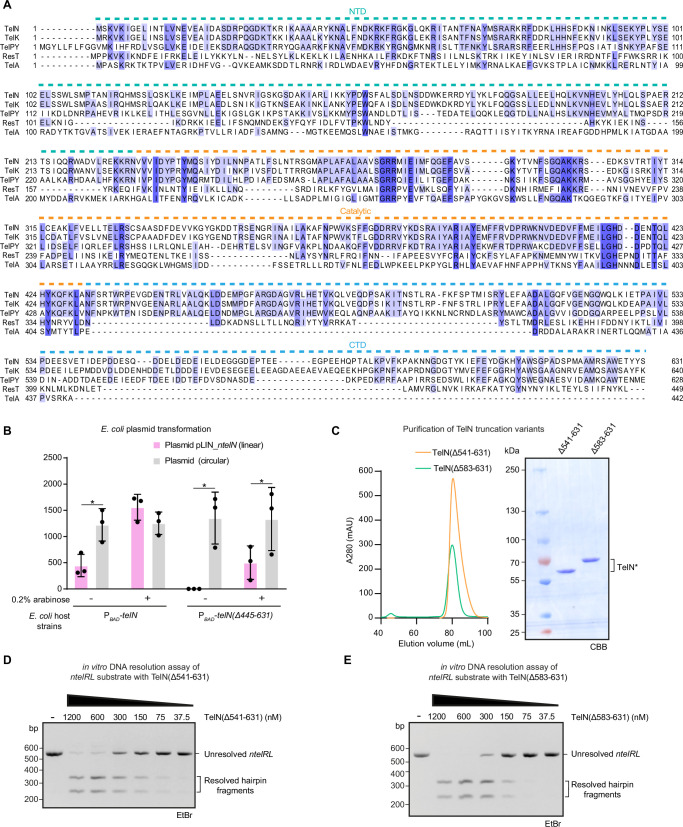 Figure 9
Figure 9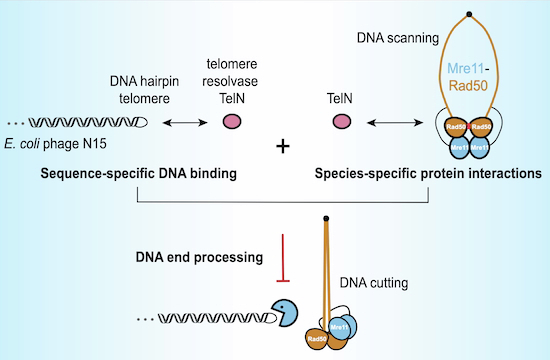 Figure 12
Figure 12 Figure 13
Figure 13- —Swiss National Science Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsDNA Repair Mechanisms · CRISPR and Genetic Engineering · Bacterial Genetics and Biotechnology
Introduction
Genome integrity is essential for survival and propagation. For organisms with linear chromosomes, maintaining genomic termini presents two fundamental challenges across all domains of life. First, semi-conservative replication fails to fully duplicate chromosome termini, known as the end replication problem, resulting in the need for dedicated enzymes such as telomerase to prevent progressive sequence loss. Second, the ends of linear chromosomes must be protected from inappropriate processing by DNA repair machinery, referred to as the end protection problem. To address these fundamental challenges, eukaryotic cells have evolved specialized nucleoprotein structures called telomeres, to prevent failures in end replication and protection leading to nucleolytic degradation, chromosome end-to-end fusion, and ultimately cell cycle arrest or cell death (De Lange, 2009; Lazzerini-Denchi and Sfeir, 2016).
While most bacterial and archaeal chromosomes are circular, lacking DNA ends, some prokaryotic organisms harbor linear replicons, requiring protective mechanisms to preserve genome integrity. Bacterial telomeres fall into two distinct categories (Chaconas and Kobryn, 2010; Volff and Altenbuchner, 2000). The first type comprises terminal inverted repeat (TIR) sequences bound by terminal proteins, as found, for instance, in Streptomyces (Bao and Cohen, 2001) and in the Bacillus subtilis phage phi29 (Yoshikawa and Ito, 1981). The other category consists of covalently closed hairpin DNA ends, identified in diverse organisms, including the Lyme spirochete Borrelia burgdorferi (Fraser et al, 1997), the plant pathogen Agrobacterium tumefaciens (Goodner et al, 2001) as well as in various bacteriophages such as Klebsiella oxytoca phage phiKO2 (Stoppel et al, 1995) and E. coli phage N15 (Rybchin and Svarchevsky, 1999).
The mechanism of hairpin telomere replication has been extensively studied in the E. coli bacteriophage N15. The N15 prophage does not integrate into the host bacterial genome but exists as a linear DNA molecule with covalently closed hairpin ends. During N15 replication, the telomere resolvase TelN linearizes a dimeric circular replication intermediate by cleaving at two specific telomeric junction sites and sealing the ends, generating two monomeric linear copies of the chromosome with covalently closed hairpin telomeres (Deneke et al, 2000; Ravin, 2003; Ravin et al, 2001). TelN cleaving-joining activity requires its cognate ntelRL sequence, a 56-bp palindromic core essential for TelN binding and catalytic activity with additional repeats found in the flanking DNA sequences (Deneke et al, 2002). TelN generates a linear DNA molecule with covalently closed hairpin ends designated ntelL (left end) and ntelR (right end), obtained from processing of ntelRR and ntelLL sites produced by DNA replication (Ravin, 2015). Notably, the N15 TelN-ntelRL system was used to linearize the 4.6 Mb E. coli circular genome, resulting in viable cells with either a single linear chromosome (Cui et al, 2007) or with two complementary linear fragments (Liang et al, 2013), harboring covalently closed hairpin telomeres. These cells exhibited normal physiology, aside from the dispensability of the chromosome dimer resolution pathway (dif, XerCD), as expected for linear chromosomes.
While TelN effectively addresses the end replication problem through hairpin telomere creation and maintenance, how these linear structures evade processing by host nucleases is unknown. A significant threat to hairpin telomeres is the conserved Mre11-Rad50 (MR) complex, a key component of cellular double-strand break DNA repair machinery (Rojowska et al, 2014; Hopfner, 2023). The MR complex (also known as SbcCD in bacteria, including E. coli) comprises a Rad50 ATPase dimer associated with an Mre11 nuclease dimer, with an additional regulatory factor in eukaryotes (Nbs1 in mammals or Xrs2 in yeast) forming the MRN/MRX complex. MR specifically discriminates and processes linear DNA through a conserved mechanism in which Rad50 senses DNA ends, triggering ATP-dependent conformational changes that activate Mre11 nuclease activity (Käshammer et al, 2019); (Fig. EV1A). This enables both strand-specific endonucleolytic and 3′-5′ exonucleolytic cleavage at diverse DNA end structures, including free termini, protein-blocked ends, and hairpins (Connelly et al, 1998, 1999, 2003; Eykelenboom et al, 2008; Gut et al, 2022; Paull, 2018; Saathoff et al, 2018).
The MR complex, though essential for DNA repair, must be tightly regulated at telomeres. In eukaryotes, multiple parallel mechanisms protect chromosomal termini. Mammalian telomeres form compact T-loop structures through the shelterin complex, sequestering DNA ends (Van Ly et al, 2018; Smith et al, 2020), while in budding yeast, multiple Rap1 proteins sterically bind and cap telomeric regions, presumably stiffening DNA (Le Bihan et al, 2013). Simultaneously, three evolutionarily distinct peptide motifs are known to inhibit MRN/MRX directly. The iDDR motif in mammalian shelterin protein TRF2, MIN in telomere-associated Taz1 (fission yeast), and BAT in telomere-associated Rif2 (budding yeast), all target the same β-sheet of RAD50 to block nucleolytic processing at chromosome ends (Bombarde et al, 2010; Fan et al, 2025; Khayat et al, 2021, 2024; Marsella et al, 2021; Myler et al, 2023; Okamoto et al, 2013; Roisné-Hamelin et al, 2021).
Similar protection strategies against MR are needed in prokaryotes with linear replicons but remain largely uncharacterized. Hairpin DNA is efficiently processed by the MR complex (Saathoff et al, 2018); thus, a mechanism for hairpin telomere protection from inadvertent degradation must be in place for N15 propagation. In this study, we discover a novel role for TelN in hairpin telomere protection from E. coli MR processing, which depends on both sequence-specific DNA binding and species-specific protein–protein interactions. This protective function is distinct from TelN’s previously known catalytic activity and independent of its C-terminal domains. Our data suggest that MR inhibition at telomeres may represent one of the earliest evolutionary solutions to the challenge of maintaining linear chromosomes, establishing fundamental principles of telomere protection conserved from bacteria to humans.
Results
Chromosome linearization is lethal in the presence of host DNA repair nuclease Mre11-Rad50 in B. subtilis
To assess whether bacterial chromosomes can be linearized across different species, we attempted to generate linear chromosomes in B. subtilis using the phage-encoded telomere resolvase TelN and its ntelRL core sequence extended by neighboring sequences from the N15 phage (Fig. 1A), an approach previously shown to successfully linearize the E. coli chromosome (Cui et al, 2007). We therefore engineered a B. subtilis strain containing this ntelRL sequence, denoted as WT^ntelRL^. Despite multiple attempts, no clones carrying both telN and intact ntelRL could be obtained (Figs. 1B and EV1B), with rare suppressor clones carrying various deletions or mutations in the ntelRL insertion site (Fig. EV1C). We wondered whether the generated linear chromosomes might be unstable due to their targeting by host DNA repair nucleases. To test this, we repeated the experiment in a strain lacking a gene for Mre11-Rad50 (ΔMR^ntelRL^). Strikingly, transformation with a xylose-inducible telN construct yielded significantly more colonies in the ΔMR^ntelRL^ strain compared to WT^ntelRL^, where only a few, small-sized colonies were observed (Figs. 1B and EV1B). These results suggest that MR actively prevents the formation or maintenance of linear chromosomes in B. subtilis, but not in E. coli (Cui et al, 2007).Figure 1. Antagonistic roles of Mre11-Rad50 and TelN in maintenance of linear DNA with hairpin ends.(A) Schematic depiction of chromosome linearization in B. subtilis. Upon induction of TelN expression with xylose (P_xyl-telN), TelN recombines ntelRL inserted at yoxD using its cleaving-joining activity, resulting in a linear B. subtilis chromosome with hairpin termini. The formation of these hairpin telomeres locally alters gene expression, resulting in reduced spectinomycin resistance (red arrow). (B) Counts of erythromycin-resistant colonies for the indicated B. subtilis backgrounds (WT^ntelRL^ and ΔMR^ntelRL^) after transformation with telN (Pxyl-telN::ermB) or smc (smc::ermB) constructs. Data were normalized to the ΔMR^ntelRL^ strain (set as 100%). Means and standard deviations obtained from three biological replicates are shown. (C) Serial spot dilutions (10⁰–10⁻⁵) of B. subtilis ΔMR strains carrying either a circular or linear chromosome: ΔMR, ΔMR^ntelRL^, and ΔMR^ntelRL^TelN. Cells were spotted on nutrient agar (ONA) plates supplemented as indicated. Growth was assessed under three conditions: ONA, ONA + 0.5% xylose, and ONA + spectinomycin 50 μg/mL + 0.5% xylose. (D) Expression of TelN promotes transformation with linear pLIN plasmid. Counts of chloramphenicol-resistant colonies for the indicated E. coli backgrounds after transformation with either a linear plasmid with ntel hairpin ends (ntelL and ntelR), pLINntelN, or a control circular plasmid. Strains were grown with (+) or without (−) 0.2% arabinose. WT: wild-type strain, PBAD: WT strain with chromosomal integration of arabinose-inducible promoter alone, PBAD-telN: WT strain with chromosomal integration of arabinose-inducible TelN, PBAD-telN(Y425F): WT strain with chromosomal integration of arabinose-inducible TelN(Y425F). Each transformation from three biological replicates was plated separately, and colonies were counted as the experimental unit. Sample size was chosen based on prior experience with similar assays, where three independent replicates consistently captured the variability in transformation efficiency. Colony counts from replicate plates were used to calculate the mean and standard deviation. Asterisks indicate samples (circular vs linear) with a p value, obtained by paired t-tests, lower than 0.05. WT: p = 0.0073; P_BAD without arabinose: p = 0.0245; P_BAD_ with arabinose: p = 0.019. Note that data for selected samples are also shown in Fig. 3B. (E) Counts of chloramphenicol-resistant colonies for the indicated E. coli backgrounds after transformation with either pLIN_ntelN or a control circular plasmid. Means and standard deviations from three biological replicates are shown. Asterisks indicate samples (circular vs linear) with a p value, obtained by paired t-tests, lower than 0.05. WT: p = 0.009; Δrad50: p = 0.0471; Δmre11: p = 0.0144; ΔmutS: p = 0.0372.
To confirm and further characterize linear chromosome formation in B. subtilis, we first used antibiotic sensitivity as a proxy for genomic linearization, employing a spectinomycin resistance gene inserted at ntelRL which becomes sensitized to the DNA hairpin formation likely due to loss of DNA super helicity near the end (Fig. 1A). We observed two distinctive growth phenotypes in B. subtilis strains with linearized chromosomes. First, strains with a linear chromosome displayed impaired growth and the frequent appearance of suppressors in the absence of xylose, likely due to a requirement of TelN for telomere resolution (Fig. 1C). Second, these strains showed reduced spectinomycin resistance, consistent with lowered gene expression at the DNA end, likely due to loss of DNA underwinding (Fig. 1C), as demonstrated for linear versus circular plasmids in Borrelia burgdorferi where topological changes directly affect transcriptional activity (Beaurepaire and Chaconas, 2007). Moreover, we performed Southern blotting to confirm the linearization of ntelRL DNA on the B. subtilis chromosome (Fig. EV1D,E). Taken together, these data provide strong evidence for successful chromosome linearization in B. subtilis strains lacking Mre11-Rad50 but not in the wild type.
TelN is required for the protection of hairpin telomeres in E. coli
These results identify MR as a fundamental barrier to linear chromosome formation or maintenance in B. subtilis. However, the successful linearization of E. coli chromosomes using the same TelN-ntelRL system (Cui et al, 2007) suggests that in E. coli, this barrier does not exist or that it is overcome by dedicated mechanisms. We therefore next investigated how E. coli stably maintains linear DNA molecules despite the presence of MR. As a simple model system, we chose the phage N15-derived linear plasmid (previously commercialized under the name pJAZZ), here denoted as pLIN_ntelN (Godiska et al, 2009). pLIN_ntelN carries its own copy of the telN gene necessary for its replication as well as a chloramphenicol-resistant marker for selection of transformants (Fig. EV1F). We first assessed the transformation efficiency of E. coli host cells containing or lacking an additional telN gene. Briefly, telN was placed under an arabinose-inducible promoter (P_BAD_) and integrated into the E. coli K12 chromosome near the glmS locus by mini-Tn7 transposon-based insertion (Bao et al, 1991). Strikingly, in the absence of chromosome-encoded telN, transformation of linear DNA was severely impaired compared to a circular control plasmid (Fig. 1D), consistent with previous observations that linear DNA with hairpin telomeres transforms E. coli with ~100-fold lower efficiency than circular DNA (Dorokhov et al, 2004). However, transformation of linear and circular DNA was similarly efficient when TelN was already present in host cells prior to transformation (Fig. 1D), consistent with the previous observation that the presence of the N15 prophage or of TelN-expressing plasmids increases transformation efficiency with linear DNA (Dorokhov et al, 2004). The presence of host-encoded telN is therefore needed for efficient uptake or maintenance of the linear plasmid with hairpin telomeres, presumably until plasmid-encoded TelN accumulates to sufficient levels.
Based on our previous findings in B. subtilis (Fig. 1B), we suspected that the E. coli MR may be responsible for the low transformation efficiency of pLIN_ntelN in the absence of host-encoded telN. Thus, we transformed both linear and circular plasmids into E. coli strains deficient in the rad50 or mre11 genes. We observed that the absence of the rad50 or mre11 genes resulted in increased transformation efficiency of pLIN_ntelN, almost to the level obtained by transformation with a circular plasmid, even in the absence of host-encoded telN (Fig. 1E). This is not the case when deleting an unrelated gene, such as mutS, in the host strain (Fig. 1E). These results demonstrate that MR is indeed responsible for the low transformation efficiency of pLIN_ntelN and suggest that TelN protects linear DNA from MR-mediated degradation in E. coli.
Inhibition of Mre11-Rad50 by TelN in vitro
To determine whether TelN inhibits Mre11-Rad50 nuclease activity in the absence of other factors, we developed an in vitro DNA degradation assay with a fluorescently labeled linear DNA substrate with a hairpin end. This substrate was generated by PCR amplification of the ntelRL sequence, along with adjacent N15 phage genomic regions, using a 5′ biotinylated primer labeled with fluorescein (Fig. 2A). TelN-mediated resolution of the ntelRL PCR product yielded fragments with one hairpin end. The fluorescently labeled hairpin substrate was immobilized on streptavidin-coated magnetic beads, using the physical size of the beads to sterically protect the biotin-labeled non-hairpin end and thus allowing MR to process the DNA from only one side, mimicking its suspected activity at unprotected telomeres (Fig. 2A).Figure 2. TelN directly inhibits Mre11-Rad50 nuclease activity independently of its catalytic function.(A) Schematic representation of the generation of a fluorescent hairpin DNA substrate for in vitro protection assays. (B,** C**) In vitro protection assay with E. coli Mre11-Rad50 using (B) TelN WT or (C) TelN(Y425F). DNA degradation products were analyzed by DNA-denaturing PAGE after biotin/SDS elution. Lower panel: representative in-gel fluorescence analysis showing DNA degradation profile. Upper panel: same gel image with reduced exposure optimized to visualize remaining substrate bands. (D) DNA binding analysis by fluorescence anisotropy. Upper panel: binding curves of TelN WT with 40 bp ntelR DNA hairpin (“hp”) and TelN(Y425F) with hp and with 80 bp ntelRR DNA duplex (“du”). Lower panel: calculated dissociation constants (KD) for different DNA substrates and TelN variants. Data points and error bars represent means and standard deviations from two technical replicates (n = 2).
We added purified MR and TelN protein (Fig. EV2A,B) to the bead-immobilized hairpin substrate and observed that in the absence of TelN, MR efficiently degraded the DNA in the presence of ATP (Figs. 2B and EV3A,B). Interestingly, the addition of TelN inhibited MR nuclease activity in a dose-dependent manner (Figs. 2B and EV3A,B). This demonstrates that TelN is sufficient to protect hairpin telomeres from E. coli MR degradation.
Hairpin telomere protection is independent of TelN’s ability to resolve hairpin telomeres
Given that TelN is primarily characterized as a telomere resolvase, we next investigated whether its catalytic activity was required for hairpin telomere protection. We thus generated a catalytically inactive TelN(Y425F) mutant using site-directed mutagenesis (Deneke et al, 2000) (Fig. EV2B). As expected, purified TelN(Y425F) showed no detectable DNA resolution activity on the ntelRL target sequence even at high protein concentrations, while TelN WT efficiently resolved the substrate (Fig. EV2C,D). Strikingly, we observed that TelN(Y425F) inhibited MR-mediated hairpin degradation as effectively as TelN WT in our in vitro protection assay (Figs. 2C and EV3C). This suggests that TelN’s resolvase and protection activities can be separated. To confirm these observations in a cellular context, we expressed TelN(Y425F) from the E. coli chromosome and assessed its impact on telomere protection. Interestingly, TelN(Y425F) improved the transformation efficiency of linear plasmid with hairpin ends similar to WT (Fig. 1D). This result further supports the notion that TelN’s protective function is mechanistically distinct from its telomere resolution activity.
DNA end binding by TelN
To understand how TelN mediates protection, we first assessed its DNA binding properties by fluorescence anisotropy. We designed fluorescein-labeled DNA substrates containing the ntelR sequence in two conformations: a 40-bp ntelR DNA hairpin and an 80-bp linear ntelRR DNA duplex (Fig. EV3D). TelN bound to these substrates with moderate affinity (dissociation constant (KD) in the ~200–300 nM range), regardless of the DNA structure or length (Fig. 2D). Due to the catalytic activity of TelN, its binding to the linear 80 bp ntelRR duplex DNA substrate could be affected by the resolution of the DNA. As expected, no significant differences in binding affinity were observed between TelN WT and TelN(Y425F) on the 40 bp ntelR hairpin substrate, suggesting that the catalytic activity of TelN does not influence its DNA binding properties to a single binding site (Fig. 2D). Surprisingly, the binding affinity of TelN(Y425F) remained comparable between the hairpin with a single binding site and the DNA duplex with two binding sites, indicating a lack of strong cooperativity in DNA binding and implying that two TelN proteins bind largely individually to the two binding sites (Fig. 2D). We conclude that TelN recognizes its target sequence with moderate affinity. Our findings suggest that TelN binds to the educt and the product of the enzymatic reaction with similar affinity, implying that it remains bound to the target DNA after telomere resolution, unlike typical enzymes. This behavior is consistent with observations with related telomere resolvases: ResT from B. burgdorferi binds hairpin telomeres and can promote telomere fusion, the reverse reaction (Kobryn and Chaconas, 2005), structural studies reveal an intact dimer of TelA binding to hairpin products (Shi et al, 2013), and TelK shows limited turnover on hairpin DNA products (Huang et al, 2004).
The C-terminal domains of TelN are dispensable for hairpin telomere protection
The C-terminal domains of telomere resolvases are less well conserved across species, and the functions remain unknown (Fig. EV4A). To investigate their role in TelN’s protective function, we generated three truncation variants lacking the C-terminal domain(s): Δ445–631, Δ541–631, and Δ583–631 (Fig. 3A). We integrated these mutants into the E. coli chromosome under arabinose-inducible control to assess their ability to protect linear DNA in vivo. TelN WT provided protection even without induction. All truncation variants, including the deletion of both C-terminal domains (Δ445–631), protected linear DNA with hairpin ends but required arabinose induction to restore the transformation efficiency of linear plasmids to (near) wild-type levels (Figs. 3B and EV4B).Figure 3. The C-terminal domains of TelN are dispensable for telomere protection.(A) Left panel: Domain organization of TelN with positions of truncations (Δ445–631, Δ541–631, and Δ583–631). Right panel: Alphafold3 prediction of a TelN monomer (pTM = 0.74) showing N-terminal domain in green (NTD), catalytic domain in orange, and C-terminal domains in blue (CTD1 and CTD2). (B) Counts of chloramphenicol-resistant colonies for the indicated E. coli strains after transformation with pLIN_ntelN or a control circular plasmid. Strains were grown with (+) or without (−) 0.2% arabinose. P_BAD-telN(Δ541–631): WT strain with chromosomal integration of TelN(Δ541–631), PBAD-telNΔ583–631: WT strain with chromosomal integration of arabinose-inducible TelN(Δ583–631). Means and standard deviations from three biological replicates are shown. Asterisks indicate samples (circular vs linear) with a p value, obtained by paired t-tests, lower than 0.05. WT: p = 0.0073; P_BAD without arabinose: p = 0.0245; PBAD with arabinose: p = 0.019; PBAD-telN(Δ583–631) without arabinose: p = 0.0370; P_BAD_-telN(Δ583–631) with arabinose: p = 0.0305. Same experiment as in Fig. 1D, displaying additional data for telN truncation mutants and controls. A similar experiment using TelN(Δ445–631) is shown in Fig. EV4B. (C,** D**) DNA protection using purified TelN(Δ541–631) protein (C) and TelN(Δ583–631) protein (D). DNA degradation products were analyzed by DNA-denaturing PAGE. Lower panel: representative in-gel fluorescence analysis showing DNA degradation profile. Upper panel: same gel image with reduced exposure optimized to visualize remaining substrate bands.
We also tested whether some of these truncated variants could protect hairpin telomeres from MR degradation in vitro. Purified Δ541–631 and Δ583–631 proteins (Fig. EV4C) inhibited MR nuclease activity comparably to wild-type TelN (Figs. 3C,D and EV5A,B), while retaining full catalytic activity in telomere resolution assays (Fig. EV4D,E). These results demonstrate that the C-terminal sequences are dispensable for telomere protection, suggesting that the core protection mechanism resides in the more conserved region of the protein.
Sequence specificity of TelN-mediated protection is modulated by cellular context
To elucidate the molecular requirements for TelN-mediated protection, we tested whether binding of TelN to ntelRL was important for protection. We first tested in vivo whether TelN could protect non-cognate DNA sequences by comparing it with TelPY, a related telomere resolvase from Yersinia enterocolitica bacteriophage PY54 (Hertwig et al, 2003) (Fig. EV4A). We designed linear plasmids with two different hairpin ends for analysis (Fig. 4A). Starting from pLIN_ntelN, we produced a circular form upon inactivation of the plasmid-encoded telN gene through an early stop codon and in the same cloning step, ligating ntelR and ntelL to recreate ntelRL. We then replaced the ntelRL recognition sequence by pytelRR, the recognition sequence of TelPY, which shares 59.5% sequence identity with ntelRL (Fig. EV6A). This plasmid was transformed into a ΔMR E. coli strain expressing chromosomal telPY, yielding a linear plasmid with two pytelR hairpin ends (Fig. 4A).Figure 4. Sequence specificity of TelN-mediated DNA hairpin protection.(A) Overview of the cloning procedure for generating linear plasmids pLIN_pytel from pLIN_ntelN. Step 1: telN mutagenesis to generate a circular plasmid containing the unresolved ntelRL sequence. Step 2: Replacement of ntelRL with pytelRR sequence. Step 3: TelPY-mediated linearization to generate a linear plasmid pLIN_pytel. Adapted from (Liu et al, 2022). (B) Counts of chloramphenicol-resistant colonies for the indicated E. coli strains after transformation with linear plasmids pLIN_ntelN and pLIN_pytel or a control circular plasmid. Means and standard deviations from three biological replicates are shown. Asterisks indicate samples (linear DNAs) with a p value, obtained by paired t-tests, lower than 0.05. P_BAD-telN without arabinose: p = 0.0136; PBAD_-telN with arabinose: p = 0.0469. (C) Left panel: Schematic representation of DNA degradation substrates. Three distinct fluorescently labeled DNA substrates were tested simultaneously: two fluorescently labeled ntel hairpin substrates of 329 and 237 bp, and one nonspecific linear DNA fragment of 411 bp. Right panel: Representative example of an in-gel fluorescence analysis showing DNA products. Image overlay displaying three substrates in different colors (green/blue: ntel hairpin substrates appearing at 658 and 474 bp due to denaturation; red: nonspecific linear DNA at 411 bp).
Using the two linear constructs, pLIN_ntelN with ntel and pLIN_pytel with pytel hairpin ends, we assessed protection specificity through plasmid transformation assays. Despite the sequence similarity between their respective binding sites, the proteins exclusively protected their cognate DNA sequence. Chromosomally encoded telN enabled efficient transformation of pLIN_ntelN even without arabinose induction, but failed to support transformation of pLIN_pytel (Fig. 4B) even with induction. Similarly, TelPY showed similar sequence specificity by only protecting plasmids with its cognate pytel hairpin ends, though requiring arabinose induction for protection (Fig. 4B). This strict specificity suggests that sequence-specific DNA binding is essential for protection, ruling out mechanisms that solely rely on a direct interaction between the telomere resolvase protein and the MR complex. Notably, TelPY protects (pytel) hairpin ends from E. coli MR despite its higher sequence divergence from TelN (Fig. EV4A), possibly by steric hindrance rather than specific protein–protein contacts.
To gain more insights into the DNA specificity, we examined the protective capacity of TelN under defined biochemical conditions. We assessed TelN’s ability to protect three distinct DNA substrates in vitro: two fluorescently labeled ntel hairpins (ntelL and ntelR) and a nonspecific fluorescent linear DNA fragment lacking ntelRL sequences (Fig. 4C). Interestingly, at slightly elevated protein concentrations, TelN also protected nonspecific DNA ends from MR degradation, with nearly complete protection of all substrates at 640 nM TelN (Figs. 4C and EV6B–D). This discrepancy between in vivo specificity and broader substrate protection in vitro suggests that spatial organization likely modulates TelN’s protective function in cells. While TelN can inhibit MR activity on various DNA substrates when present at high concentrations in solution, the cellular environment, with its vast excess of non-cognate DNA, likely constrains this activity through sequence-specific recruitment, ensuring protection is directed specifically to telomeric sequences, while DNA repair can occur elsewhere in the genome.
Species specificity of TelN-mediated protection
To further characterize the protection mechanism, we investigated whether TelN could inhibit MR complexes from different species. We compared TelN’s ability to inhibit nucleolytic processing by both E. coli MR and a eukaryotic counterpart, the MRX complex from S. cerevisiae. Despite their functional similarity, these complexes only share limited sequence homology. E. coli MR proteins share only limited sequence similarity with their yeast counterparts. Using a fluorescently labeled hairpin substrate immobilized on streptavidin-coated magnetic beads, we compared TelN’s protective activity against purified E. coli MR versus its eukaryotic ortholog MRX. TelN effectively blocked E. coli MR nuclease activity, while showing no protection against yeast MRX degradation (Fig. 5A,B). This species-specificity suggests that TelN makes specific contacts with the bacterial MR complex, pointing toward a protection mechanism involving direct protein–protein interactions with the E. coli complex, thus providing an explanation for the lack of protection observed in B. subtilis. Alternatively, or additionally, telomere resolvases may hinder bacterial MR by steric hindrance but not yeast MRX, for instance, due to the greater length of its coiled coils.Figure 5. TelN-mediated protection exhibits species specificity.(A) Hairpin DNA degradation by S. cerevisiae MRX complex. Lower panel: representative in-gel fluorescence analysis showing DNA degradation profile after incubation with pSae2 and MRX. Upper panel: same gel image with exposure optimized to visualize remaining substrate bands. (B) DNA protection by TelN from degradation of E. coli MR complex. Lower panel: representative in-gel fluorescence analysis showing DNA degradation profile after incubation with MR. Upper panel: same gel image with exposure optimized to visualize remaining substrate bands.
Discussion
Maintenance of chromosome ends and protection from cellular nucleases pose a fundamental challenge for organisms with linear chromosomes. Eukaryotes have evolved several independent mechanisms to protect their telomeres from degradation, prevent end-to-end chromosome fusions, and telomere erosion. In contrast, the mechanisms governing DNA end protection in bacteria with linear chromosomes remain poorly understood. This study demonstrates the role of phage-encoded TelN telomere resolvase in hairpin telomere protection. Using both in vivo and in vitro approaches, we show that TelN is essential and sufficient to protect hairpin telomeres from MR nuclease degradation in E. coli (Figs. 1D and 2B). Notably, the hairpin resolvase catalytic activity of host-encoded TelN is not needed for efficient protection of linear DNA with hairpin ends (Figs. 1D and 2C), revealing a clear separation of function.
Molecular mechanisms underlying TelN-mediated end protection
Our findings suggest that TelN protects chromosome ends through multiple coordinated steps. The first step involves the formation of a telomeric hairpin by TelN itself, which, in addition to solving the DNA end replication problem, eliminates free 3’ and 5’ ends that would otherwise be susceptible to exonuclease activity, such as from RecBCD. Second, our data demonstrate that sequence-specific DNA recognition and binding are required, as TelN protects only its cognate sequence from MR in vivo (Fig. 4B). Third, the specific inhibition of E. coli MR, but not yeast MRX, points to a direct protein–protein interaction between TelN and the E. coli MR complex (Fig. 5), possibly in addition to steric blockage as indicated by TelPY’s inhibition of E. coli MR. Together, these results suggest that at least two features, DNA binding and MR interaction, act in concert to inhibit MR. Loss of either component permits MR to cleave the hairpin, likely enabling further degradation by exonucleases such as RecBCD. This dual requirement mirrors strategies employed by eukaryotic telomere protection factors. For instance, in S. cerevisiae, the telomere-associated protein Rap1 recognizes, and binds repeat sequences in tandem, presumably stiffening telomeric DNA (Williams et al, 2010; Le Bihan et al, 2013). Rap1 also recruits another telomere-associated protein, Rif2, which directly inhibits MRX through its BAT motif binding to Rad50 (Roisné-Hamelin et al, 2021).
The observed difference between TelN’s sequence-specific protection in vivo and its ability to protect nonspecific DNA in vitro (Fig. 4B,C) provides important insights into the spatial regulation of telomere protection. While TelN can inhibit MR activity on various DNA substrates at elevated concentrations in solution, its protective function requires precise binding to specific sequences in a cellular context. This mirrors the eukaryotic telomere protection factor Rif2, which inhibits MRX endonuclease activity in vitro without requiring DNA recruitment (Marsella et al, 2021), yet in vivo, it is spatially constrained through Rap1-dependent localization at telomeres (Roisné-Hamelin et al, 2021). This permits the distinction between chromosome ends that need to be protected from DNA damage and sites that need to be repaired. Likely, TelN remains bound to newly generated DNA ends, thus ensuring protection of DNA ends even at low TelN concentrations.
Recent structural insights into MR conformational states during DNA end processing show that recognition of a DNA end induces a ring-to-rod transition of the coiled coils following ATP hydrolysis (Käshammer et al, 2019; Gut et al, 2022). The Mre11 dimer moves to the side, forming a nuclease proficient complex. TelN might hamper the conformational change of the MR complex, preventing ATP binding and hydrolysis, thus blocking the release of the nuclease and preventing DNA end processing. MR would be stuck in its ATP-bound state, unable to capture telomeric structures. Our in vitro assays support this hypothesis, demonstrating that TelN directly inhibits the nuclease activity of the MR complex (Fig. 2B), thereby protecting DNA ends from degradation. A similar mechanism is proposed in eukaryotic systems, where, for instance, the iDDR motif in mammalian shelterin protein TRF2, MIN in telomere-associated Taz1 (fission yeast), and BAT in Rif2 (budding yeast) inhibit MRN/X-dependent DNA resection at telomeres by binding the same exposed β-sheet region of the RAD50 ATPase head (Fan et al, 2025; Khayat et al, 2024). The structural basis of potential TelN-MR interactions remains to be determined, and high-resolution structural studies using cryo-electron microscopy could reveal whether TelN induces conformational changes in the E. coli MR complex. Our attempts to detect direct interactions through co-immunoprecipitation between TelN and MR were unsuccessful, suggesting that their interaction might be transient or context-dependent.
Higher-order structure formation in DNA end protection
Several observations suggest that TelN may induce structural changes in telomeric DNA for efficient protection. The natural target sequence of TelN appears significantly larger (310 bp) than the recognition sites required for efficient recombination (ntelRL, ntelRR, or ntelLL, 56 bp) (Deneke et al, 2002). These recognition sites contain the core telO sequence (28 bp) plus L3 and R3 sequence motifs (each 10 bp) flanking the core (Deneke et al, 2000). At least two degenerate copies of the L3/R3 motifs (L1 and L2 or R1 and R2, respectively) are found in the neighboring regions. Based on the TelK-DNA crystal structure (Aihara et al, 2007), it is conceivable that TelN’s CTD1 domain binds all these repeat motifs to recruit multiple TelN proteins and form a protective nucleoprotein assembly. However, this domain is not essential for protection (Fig. EV4B). Moreover, our in vitro data demonstrate that even at low concentrations, TelN efficiently protects cognate DNA sequences from MR degradation (Fig. 2B), which could potentially be explained by the formation of higher-order structures beyond simple DNA binding. Notably, structural analyses of related telomere resolvases, such as TelK from Klebsiella oxytoca phage phiKO2, bound to its minimal target DNA sequence (Aihara et al, 2007), reveal a basic nucleoprotein complex that would putatively readily be overcome by MR. We therefore speculate that TelN contributes to the compaction of hairpin telomeres folding into a larger structure, such as the Telomere (T)-loops observed in mammals (Van Ly et al, 2018; Smith et al, 2020). TelN-mediated DNA looping can be envisaged as an architectural solution to protect hairpin telomeres. It would be an effective mechanism to mediate end protection by sequestering and masking the extreme chromosome termini.
Evolutionary implications
Our findings provide fundamental insights into bacterial telomere biology and broaden our understanding of DNA end protection in prokaryotic systems. The dispensability of most of the TelN C-terminal domain for protection (Fig. 3), which is highly variable among telomere resolvases, raises the possibility that other bacterial telomere resolvases might share similar protective functions in the more N-terminal domains. The parallels between bacterial and eukaryotic telomere protection suggest that conserved strategies have evolved to solve the end protection problem. TelN-mediated protection of N15 hairpin ends is crucial for phage survival and propagation and is likely a result of evolutionary adaptation, providing valuable insights into phage-host coevolution. In this regard, MR is acting as a phage defense system with TelN providing counter-defense activity, in analogy to the Gam protein (gamma) of bacteriophage lambda that by protein-DNA mimicry counters the DNA repair and defense system RecBCD (and also inhibits MR/SbcCD)(Kulkarni and Stahl, 1989; Wilkinson et al, 2016). These examples illustrate convergent evolutionary strategies where distantly related phages have independently evolved distinct molecular mechanisms to counteract the same fundamental threat from the bacterial DNA repair nucleases MR and RecBCD.
Methods
Reagents and tools tableReagent/resourceReference or sourceIdentifier or catalog number Experimental models List of B. subtilis andE. coli strainsThis paperTable EV1 Recombinant DNA List of plasmidsThis paperTable EV2 Antibodies
Oligonucleotides and other sequence-based reagents Oligonucleotide sequence informationThis paperTable EV3Prime-IT II Random Primer labeling kitAgilent Technologies300385 Chemicals, enzymes and other reagents ATP solution (100 mM)Jena BioscienceNU-1010Dithiothreitol (DTT)PanReac AppliChemA1101Sodium dodecyl sulfate (SDS)Sigma-Aldrich71736D-BiotinRoth3822.4ChloramphenicolSigma-AldrichC0378Spectinomycin dihydrochloridePanReac AppliChemA3834,0005T1 Streptavidin-coated Dynabeads (Dynabeads™ MyOne™ Streptavidin T1)Thermo Fisher Scientific65601Potassium acetate 99%abcrAB118624Rubidium chloride (RbCl)Sigma-Aldrich83979Calcium chloride dihydrate (CaCl₂ ·2H₂O)Serva39551.01Manganese (II) chloride tetrahydrate (MnCl₂ ·4H₂O)Sigma-AldrichM3634Magnesium chloride hexahydrate (MgCl₂ ·6H₂O)Sigma-AldrichM2670Potassium chloride (KCl)Sigma-Aldrich60130Sodium chloride (NaCl)Sigma-Aldrich31434-1KG-RSodium hydroxide (NaOH)Sigma-Aldrich221465GlycerolSigma-Aldrich49770Trizma baseSigma-AldrichT15033-(N-morpholino) propanesulfonic acid (MOPS)Sigma-AldrichM1254Ethylenediaminetetraacetic acid (EDTA)HuberLabA4892.0500Tris(2-carboxyethyl) phosphine (TCEP)Sigma-Aldrich6465472-mercaptoethanolSigma-AldrichM-7154ImidazoleMerck1047160250HiTrap Q HP column 5 mLCytiva17115401HisTrap HP 5 mLCytiva17524802Superose 6 Increase 10/300 GLCytiva29091596HiLoad Superdex 200 columnCytiva28989335Novex 4-12% Tris-Glycine GelsLife TechnologiesXP04125BOXD-(+)-XyloseBiochemicaA2241,0500L-(+)-ArabinoseSigma-AldrichA3256AgaroseSigma-AldrichA9539AcrylamideHuberlabA0385.0500Ethidium bromide (10 mg/mL)Thermo Fisher Scientific15585011Bovine Serum Albumin (BSA)Sigma-AldrichA9418Protease Inhibitor Cocktail (PIC)Sigma-AldrichP8849Isopropyl β-d-1-thiogalactopyranoside (IPTG)Thermo Fisher ScientificR0393BsaI-HFv2New England BiolabsR3733LXhoINew England BiolabsR0146SRadiantDy™ 632 500 MOB molecular weight ladderEurogentec051023Hybond-N+ nylon membranesGE HealthcareRPN203B Software AlphaFold3Jumper et al, Evans et al, https://alphafoldserver.com/ ChimeraX V1.4Goddard et al, Pettersen et al, https://www.cgl.ucsf.edu/chimerax GraphPad Prism V10.5.0N/A https://www.graphpad.com/ SnapGene software V8.0.3N/A https://www.snapgene.com/ ImageQuant TL 1D V8.1GE Healthcare http://www.gelifesciences.com/en/us/shop/protein-analysis/molecular-imaging-for-proteins/imaging-software/imagequant-tl-8-1-p-00110 ImageJ software V1.53eNational Institutes of Health (NIH) https://imagej.nih.gov/ij
Other Original dataThis paper https://data.mendeley.com/datasets/8trn4bptpt/1
B. subtilis strain construction
All strains constructed in this work are derived from the 1A700 isolate (Table EV1). Natural competence was used to engineer strains at the yoxD rtp proH, amyE, cgeD, sbcC, and sbcD loci by allelic replacement, as described in (Diebold-Durand et al, 2019). Strains were selected on SMG-agar plates under appropriate antibiotic selection at 37 °C. Genotypes were verified for single-colony isolates by PCR and Sanger sequencing (Microsynth) as required.
Transformation assay in B. subtilis
Transformation assays were performed with naturally competent B. subtilis cells. To introduce genomic DNA, we used 60–80 ng of DNA from a strain carrying the appropriate chromosomal antibiotic resistance marker. Plates containing transformant colonies were imaged, and colonies were manually counted.
Viability assessment by dilution spotting
Bacterial strains were initially cultured on Oxoid Nutrient Agar (ONA) plates supplemented with 0.5% xylose. Single colonies were subsequently inoculated into Luria-Bertani (LB) medium containing 0.5% xylose and cultured overnight at 30 °C with continuous agitation. The overnight cultures were subjected to serial 1:10 dilutions, and bacterial density was estimated by measuring optical density at 600 nm (OD600). Fresh LB medium supplemented with 0.5% xylose was inoculated with the overnight culture to an initial OD600 of 0.001. These cultures were incubated at 37 °C until reaching the exponential phase (OD600 ≈ 0.3). For spotting assays, 200 µL of the exponential phase culture was serially diluted in a 96-well plate to achieve dilutions ranging from 10⁻¹ to 10⁻⁷. Technical duplicates of 5 µL from each dilution were spotted onto ONA plates containing selective conditions. Colony growth was documented by imaging after 19 h of incubation at 37 °C.
E. coli strain construction
A mini-Tn7 transposon carrying araC and different TelN constructs [TelN WT, TelN(Y425F), TelN(Δ445–631), TelN(Δ541–631), and TelN(Δ583–631)] under the P_BAD_ promoter was integrated into a neutral E. coli chromosomal locus downstream of glmS by triparental mating, as previously described (Bao et al, 1991). This approach generated multiple E. coli strains, each expressing a distinct TelN variant (Table EV1).
E. coli chemically competent cell preparation and transformation
To prepare chemically competent E. coli cells, a sterile flask containing the required volume of LB medium was inoculated with a few colonies from a freshly streaked plate and grown at 37 °C with shaking until an OD600 of 0.4–0.6 was reached. The culture was chilled on ice for 15 min and harvested by centrifugation (5000 rpm, 10 min, 4 °C). Cell pellets were gently resuspended in ice-cold TBF I buffer (30 mM potassium acetate, 100 mM RbCl, 10 mM CaCl₂, 50 mM MnCl₂*4H₂O, 15% glycerol, pH 5.8), using a volume equivalent to 0.4× the original culture volume. After 5 min incubation on ice and subsequent centrifugation, pellets were resuspended in ice-cold TBF II buffer (10 mM MOPS, 10 mM RbCl, 75 mM CaCl₂, 15% glycerol, and pH 6.5) at 0.02× the original culture volume. Following 15 min incubation on ice, the suspension was aliquoted (100 μL), flash-frozen in liquid nitrogen, and stored at –70 °C. Where indicated, 0.2% arabinose was added to the culture medium.
For transformation experiments, 1 ng of DNA (either linear plasmids with covalently closed hairpin ends or circular plasmids; Table EV2) was added to 100 μL chemically competent cell aliquots. After heat shock treatment at 42 °C for 1 min, cells were recovered in LB medium for 1 h at 37 °C. Transformed cells were then plated on LB agar supplemented with chloramphenicol and, where indicated, 0.02% arabinose. Plates were incubated overnight at 37 °C. Colony counts were determined using ImageJ software, and all experiments were performed in triplicate.
We performed a paired t-test analysis to compare linear versus circular plasmid transformation efficiency and indicated the p values wherever significant (below 0.05). Given that our experimental design included multiple control conditions with known expected outcomes to validate assay performance, rather than independent exploratory comparisons, we report uncorrected p-values as the primary analysis. The inclusion of multiple controls with predictable outcomes reduces the likelihood of false positive interpretations.
DNA substrate preparation and fluorescent anisotropy assay
Two DNA substrates were used for fluorescence anisotropy measurements; both derived from an 80-nucleotide palindromic oligonucleotide comprising the TelN target sequence ntelR followed by its reverse complement. A fluorescein (6FAM) label was attached to the 3′ end of the oligonucleotide (Table EV3):
5’-aattacggaacatatcagcacacaattgcccattatacgcgcgtataatgggcaattgtgtgctgatatgttccgtaatt-[6FAM]-3’
For the 40 bp specific ntelR hairpin substrate, the oligonucleotide was subjected to intramolecular annealing in buffer (100 mM NaCl, 10 mM Tris-HCl, pH 8, and 1 mM EDTA) by heating to 95 °C for 1 min, followed by rapid cooling on ice. The 80 bp specific ntelRR duplex DNA was prepared using the same oligonucleotide through intermolecular annealing at 25 °C overnight in the same buffer. Fluorescence anisotropy measurements were performed in a reaction buffer containing 25 mM Tris, pH 7.5, 50 mM KCl, 5 mM MgCl₂, and 1 mM MnCl₂. A series of TelN protein concentrations, ranging from 0 to 9.6 μM, were incubated in the presence of 50 nM fluorescein-labeled DNA substrates for 30 min at room temperature to attain equilibrium. Measurements were recorded using a Synergy Neo Hybrid Multi-Mode Microplate reader equipped with appropriate fluorescence polarization filters. Assays were conducted in black 96-well flat-bottom plates at 25 °C. Data analysis and curve fitting were performed using GraphPad Prism 10 software using non-linear regression to determine binding parameters.
Purification of the Mre11-Rad50 protein complex
Mre11-Rad50 was purified as described in (Roisné-Hamelin et al, 2024). The Mre11-Rad50 protein complex was expressed in E. coli BL21, using a dual vector system (N-terminal 10His-TwinStrep-3C tagged Mre11 and untagged Rad50). Cells were grown in TB medium at 37 °C until OD600 = 1, cooled to 18 °C, and protein expression was induced with 0.5 mM IPTG for 16 h. Cells were harvested and resuspended in lysis buffer (50 mM Tris, pH 7.5, 300 mM NaCl, 5% glycerol, 25 mM imidazole) containing 100 µL of protease inhibitor cocktail and 5 mM β-mercaptoethanol. Cells were lysed by sonication on ice, using a VS70T probe mounted on a SonoPuls unit (Bandelin), at 40% output power for 13 min with pulsing (1 s on/1 s off). After sonication and ultracentrifugation (40,000 g, 45 min), the clarified lysate underwent a three-step chromatographic purification. First, metal affinity chromatography using a HisTrap HP 5 mL column with imidazole gradient elution (25–500 mM); second, anion exchange chromatography on a HiTrap Q 5 mL column eluted with a NaCl gradient (50–1000 mM); and finally, a size-exclusion chromatography on a Superose 6 Increase 10/300 GL column equilibrated with 20 mM Tris-HCl pH 7.5, 250 mM NaCl, and 1 mM TCEP. The peak fractions containing the intact complex were concentrated to ~2.3 mg/mL, flash-frozen in liquid nitrogen, and stored at –70 °C.
Purification of TelN constructs
TelN constructs [TelN WT, TelN(Y425F), TelN(Δ541–631), TelN(Δ583–631)] with N-terminal His-TwinStrep-3C tags were expressed in E. coli BL21 cells. Cultures were grown in TB medium at 37 °C until OD600 = 1, cooled to 20 °C, and protein expression was induced with 0.5 mM IPTG for 16 h. Cells were harvested by centrifugation and resuspended in lysis buffer (50 mM Tris, pH 7.5, 300 mM NaCl, 5% glycerol, 25 mM imidazole) supplemented with 100 µL of protease inhibitor cocktail and 5 mM β-mercaptoethanol. Following sonication and ultracentrifugation (40,000 × g, 45 min), the clarified lysate underwent a four-step purification process. First, proteins were purified by affinity chromatography using a HisTrap HP column with imidazole gradient elution (25–500 mM). Then, for TelN WT, TelN(Y425F), and TelN(Δ583–631), overnight tag cleavage was performed during dialysis using 20 mM Tris, pH 7.5, 100 mM NaCl, and 5 mM β-mercaptoethanol supplemented with 500 μL 3C protease. For TelN Δ541–631, tag cleavage was performed overnight without dialysis to prevent precipitation. Finally, proteins were subjected to anion exchange chromatography using a HiTrap Q column with NaCl gradient elution. Finally, size-exclusion chromatography was performed on a HiLoad Superdex 200 column equilibrated with 10 mM Tris, pH 7.5, 200 mM NaCl, 0.1 mM EDTA, and 1 mM DTT. Purified proteins were concentrated to 7.4 mg/mL TelN WT, 7.0 mg/mL TelN(Y425F), 4.0 mg/mL TelN(Δ583–631), and 4.3 mg/mL TelN(Δ541–631), flash-frozen in liquid nitrogen, and stored at −70 °C.
DNA resolution assay
About 37 nM of 566 bp ntelRL DNA duplex, generated by PCR amplification, was incubated with increasing concentrations of TelN proteins (0–1200 nM) in 20 µL reactions containing nuclease buffer (25 mM Tris-HCl, pH 7.5, 50 mM KCl, 5 mM MgCl₂, 1 mM MnCl₂, 0.1 mg/mL BSA, and 1 mM DTT). Samples were incubated at 30 °C for 30 min, followed by heat inactivation at 75 °C for 5 min. The resulting DNA species were resolved by electrophoresis on 1.5% agarose gels containing ethidium bromide.
DNA end protection assay
A 566 bp ntelRL sequence (ntelRL sequence with additional neighboring sequences from N15 phage; Table EV3) was incubated with TelN, generating two hairpin products of 237 and 329 bp using its cleaving-joining activity. The biotinylated and fluorescently labeled 237 bp hairpin substrate was used for subsequent analyses. DNA immobilization was performed using T1 streptavidin-coated Dynabeads (10 µL, 100 µg per reaction). Beads were first pre-equilibrated using 10 volumes of nuclease buffer (25 mM Tris-HCl, pH 7.5, 50 mM KCl, 5 mM MgCl₂, 1 mM MnCl₂, 0.1 mg/mL BSA, and 1 mM DTT). About 37 nM of biotinylated hairpin DNA was bound to the beads in a 20 µL reaction at 25 °C for 15 min with constant shaking at 850 rpm. Protection assays were conducted by incubating the immobilized hairpin DNA with Mre11-Rad50 tetramer (125 or 500 nM) and increasing concentrations of TelN variants (0–1200 nM). Reactions were performed in nuclease buffer supplemented with 1 mM ATP at 37 °C for 15 min with shaking. Reactions were stopped by adding 80 µL buffer containing 10 mM EDTA and 20 mM Tris. Protected DNA was eluted by adding 100 µL preheated nuclease buffer supplemented with 25 mM biotin and 0.1% SDS at 70 °C for 15 min. The supernatant containing the eluate was separated from the beads using a magnetic rack. A RadiantDy™ 632 500 MOB molecular weight ladder (Eurogentec, LOT# 051023) was included for size determination. Samples were resolved on DNA-denaturing 5% polyacrylamide gels prepared in 1× alkaline buffer (50 mM NaOH and 1 mM EDTA). Gels were processed under light-protected conditions and subsequently scanned using a Typhoon fluorescence imager (GE Healthcare) with dual-channel acquisition: Cy2 channel for fluorescently labeled DNA substrates and Cy5 channel for ladder detection using auto PMT settings for both channels.
For species specificity experiments, 1 nM of fluorescently labeled hairpin DNA substrate was incubated with either 80 nM E. coli MR or 80 nM S. cerevisiae MRX complemented with 320 nM pSae2. Both MRX and pSae2 were kindly provided by the Cejka lab. TelN was added at three different concentrations (20, 80, and 320 nM) to assess protection against both complexes. The reactions were performed and analyzed following the same protocol described above.
For multi-substrate protection assays, we used three different fluorescently labeled DNA substrates simultaneously: a 329 bp ntel-containing hairpin labeled with TAMRA, a biotinylated 237 bp ntel-containing hairpin labeled with fluorescein and immobilized on streptavidin beads, and a 411 bp nonspecific linear DNA fragment (random sequence, non-ntelRL, non-hairpin DNA) labeled with ATTO 680. The total DNA concentration was maintained at 30 nM across all experiments, with equimolar amounts of each substrate (10 nM each). Protection assays were conducted by incubating the DNA substrates with 500 nM Mre11-Rad50 tetramer and increasing concentrations of TelN WT (0–640 nM). Reactions were performed in nuclease buffer supplemented with 1 mM ATP at 37 °C for 15 min with shaking, followed by the same stopping and elution procedures described above. For each fluorophore (fluorescein, TAMRA, and ATTO 680), the Typhoon fluorescence imager (GE Healthcare) was set with appropriate excitation and emission filter settings. For overlay images, individual fluorescence channels were pseudo-colored (fluorescein: blue, TAMRA: green, and ATTO 680: red) and combined using Adobe Photoshop.
Southern blot analysis
Genomic DNA was extracted from B. subtilis strains using standard methods and digested with BsaI and XhoI restriction enzymes (New England Biolabs) overnight at 37 °C. Digested DNA was separated on 0.8% agarose gels and transferred to nylon membranes (Hybond-N+, GE Healthcare) using alkaline transfer methods. A ^32^P-labeled DNA probe targeting the ntelRL-spectinomycin resistance gene region was hybridized overnight at 65 °C. Membranes were washed with 2× SSC/0.1% SDS at room temperature and 0.1× SSC/0.1% SDS at 65 °C, then exposed to a phosphor image screen and subsequently scanned on a Typhoon scanner (GE Healthcare). Fragment sizes were determined using DNA molecular weight standards. The analysis focused on BsaI-XhoI fragments, with expected sizes of 5797 bp for intact constructs and 4180 bp + 1621 bp for TelN-cleaved linearized chromosomes.
Supplementary information
Table EV1 Table EV2 Table EV3 Peer Review File Expanded View Figures