Synthesis of the tetracyclic skeleton of Aspidosperma alkaloids via PET-initiated cationic radical-derived interrupted [2 + 2]/retro-Mannich reaction
Ru-Dong Liu, Jian-Yu Long, Zhi-Lin Song, Zhen Yang, Zhong-Chao Zhang

TL;DR
The paper describes a new synthetic method for building a complex tetracyclic structure found in Aspidosperma alkaloids using a radical-based reaction.
Contribution
A novel PET-initiated cationic radical-derived reaction is introduced for constructing the ABCE tetracyclic framework of Aspidosperma alkaloids.
Findings
The PET-initiated reaction enables direct construction of the ABCE tetracyclic framework.
DFT calculations identified the rate-determining step involving C19–C12 bond formation and C19–C3 bond cleavage.
The reaction mechanism involves a formal 1,3-C shift and is neither fully concerted nor stepwise.
Abstract
Natural products with topologically complex architectures are important sources in drug discovery. The pursuit of conciseness and efficiency in the total synthesis of natural products promotes continuous innovation and the development of new reactions and strategies. In this work, a PET-initiated cationic radical-derived interrupted [2 + 2]/retro-Mannich reaction of N-substituted cyclobutenone provided a facile approach to the direct construction of the ABCE tetracyclic framework of Aspidosperma alkaloids. DFT calculations showed that the rate-determining step of the key PET reaction involved C19–C12 bond formation and C19–C3 bond cleavage. Investigation of the bond length changes along the IRC path, spin density, and NBO analysis indicated that this process is neither strictly concerted nor stepwise, but falls in between, and involves a formal 1,3-C shift.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
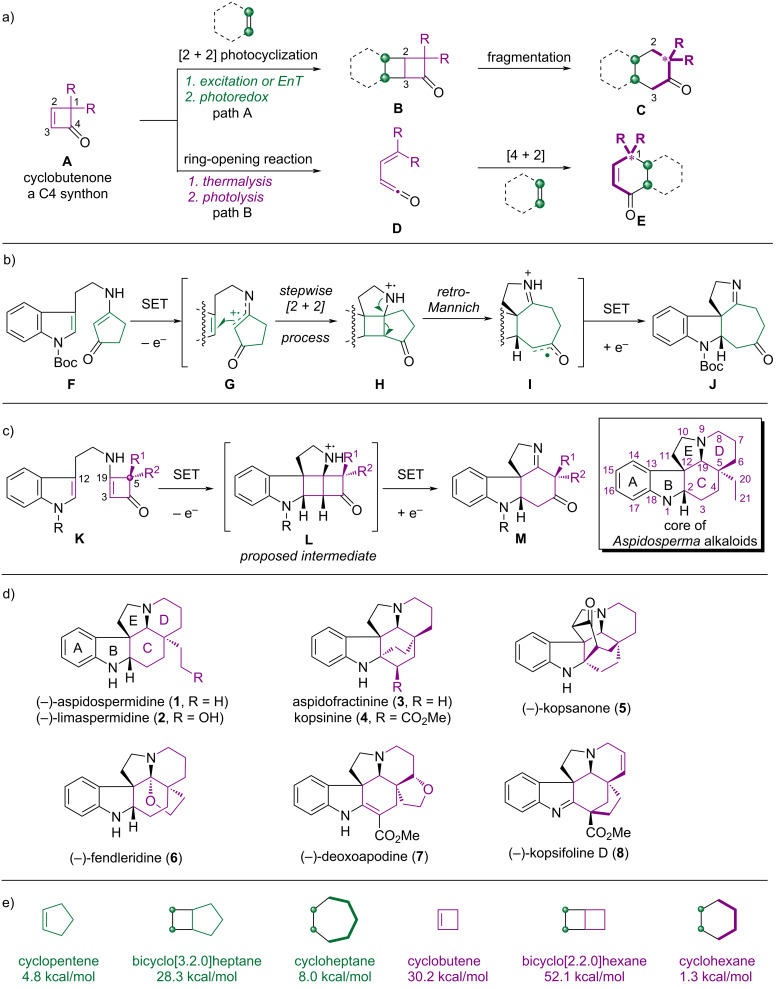 Figure 1
Figure 1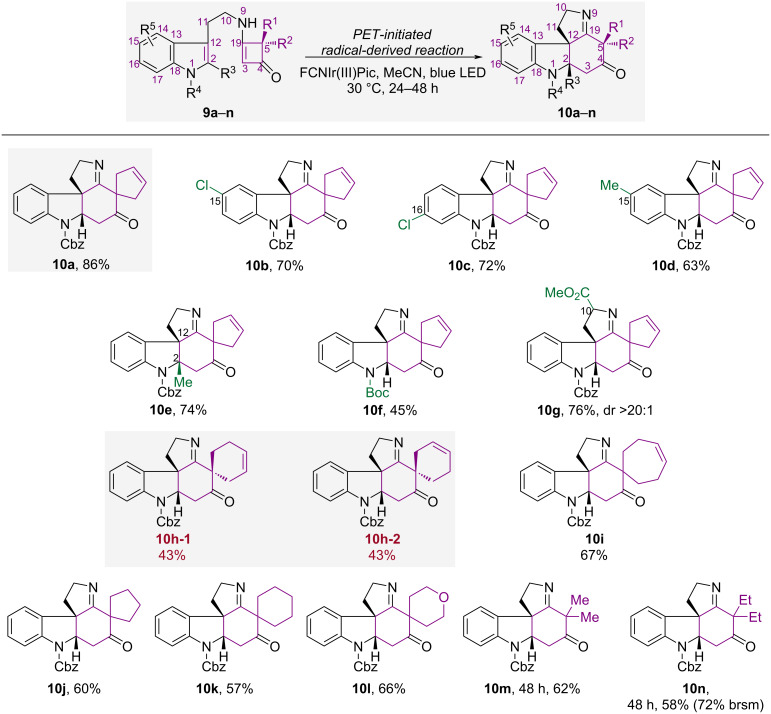 Scheme 1
Scheme 1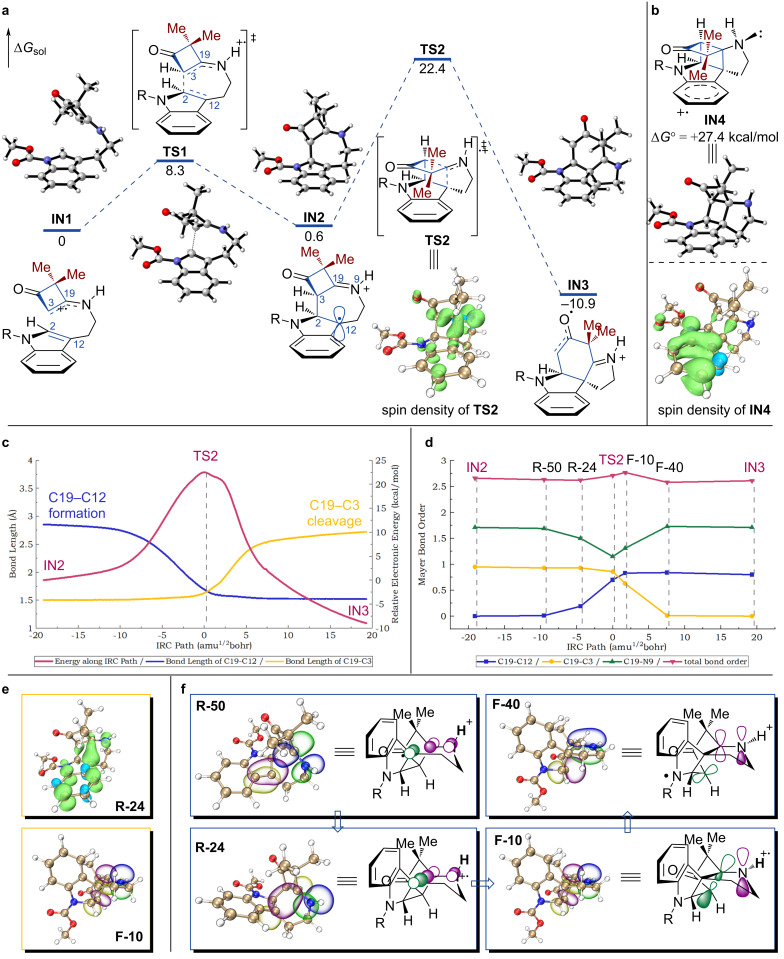 Figure 2
Figure 2|
| ||||
|
| ||||
| Entry | Photocatalyst | Solvent | Temp. | Conv./yield (%) |
|
| ||||
| 1 | FCNIrPic ( | MeCN | 30 °C | 100/86b |
| 2 | FirPic ( | MeCN | 30 °C | 83/26 |
| 3 | PhFIrPic ( | MeCN | 30 °C | 65/43 |
| 4 | in dark | MeCN | 30 °C | <5/n.d. |
| 5 | no catalyst | MeCN | 30 °C | <5/n.d. |
| 6 | FCNIrPic ( | MeOH | 30 °C | 84/23 |
| 7 | FCNIrPic ( | THF | 30 °C | 30/20 |
| 8 | FCNIrPic ( | DCM | 30 °C | 59/35 |
| 9 | FCNIrPic ( | MeCN | 30 °C | 86/42c |
| 10 | FCNIrPic ( | MeCN/PhMe | 30 °C | 67/56c,d |
| 11 | FCNIrPic ( | MeCN | 10 °C | 100/60 |
| 12 | FCNIrPic ( | MeCN | 20 °C | 100/66 |
| 13 | FCNIrPic ( | MeCN | 40 °C | 90/50 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAsymmetric Synthesis and Catalysis · Advanced Synthetic Organic Chemistry · Radical Photochemical Reactions
Introduction
Photochemical reactions, which enable the construction of topologically complex architectures from simple building blocks, have attracted considerable attention in recent decades. Numerous approaches to natural product synthesis have been reported in which a photochemical transformation represents a key step [1–3]. In this context, the photochemical [2 + 2] cycloaddition and subsequent fragmentation of the resulting cyclobutane provides a valuable strategy for synthesizing natural and unnatural products from simple building blocks [4]. Three distinct photoinitiated approaches have been established for the formation of the [2 + 2] cycloadducts: direct irradiation [5–6], energy transfer (EnT) [7], and photoinduced electron transfer (PET, or photoredox catalysis) processes [8–10].
Cyclobutenone (A) is a versatile C4 synthon [11] – its [2 + 2] photocyclization yields B, featuring a strained bicyclo[2.2.0]hexane unit [12], which can fragment to form C (Figure 1a) [13–14]. However, competitive C1–C4 bond cleavage under irradiation or heating leads to ketene D, which can undergo cycloaddition with an alkene to yield E. This fragmentation pathway dominates under various conditions (e.g., transition-metal catalysis, nucleophilic addition) and is driven by ring-strain release [11].
Synthetic plan. a) General model of cyclobutenone bond cleavage; b) our previously reported method; c) plan for indole Aspidosperma alkaloid synthesis; d) naturally occurring Aspidosperma alkaloids; e) ring strain energy of cyclopentene, bicyclo[3.2.0]heptane, cycloheptane, cyclobutene, bicyclo[2.2.0]hexane and cyclohexane.
PET, an alternative to direct excitation and EnT, enables the formation of unique radical intermediates [9–10]. We previously demonstrated the Ir-catalyzed [2 + 2] cyclization/retro-Mannich reaction of a tryptamine-substituted cyclopentenone F, which led to the formation of indoline J (Figure 1b) [15]. Unlike other reported methods [16–18], the PET reaction of F generates the cationic radical G, which initiates formation of H, which has a strained bicyclo [3.2.0]heptane core. Strain release of H triggers a downstream radical-driven retro-Mannich reaction, which ultimately results in the formation of J via reductive quenching of intermediate I.
As part of our current interest in the synthesis of complex natural products via photochemical reactions, we decided to achieve such an unusual bond cleavage (Figure 1a, path A) of cyclobutenone by generating a radical cation species via a PET reaction. The synthetic plan is shown in Figure 1c and includes a PET-initiated [2 + 2] cyclization of the tryptamine-substituted cyclobutenone K to form the radical cation L, which has a highly functionalized and rigid bicyclo[2.2.0]hexane core. Fragmentation of the C3–C19 bond would afford a redox-active intermediate which upon further reductive quenching would lead to the tetracyclic indoline M, which was expected to serve as a common intermediate for the total synthesis of Aspidosperma alkaloids. These alkaloids constitute a large family of structurally complex compounds, which incorporate a pentacyclic ABCDE skeleton (Figure 1d, 1–8) [19–23]. However, the formation of intermediate L is challenging because its ring-strain energy (Figure 1e, 52.1 kcal/mol) is higher than that of its counterpart, i.e. the bicyclo[3.2.0]heptane motif (28.3 kcal/mol) in H [24].
Herein, we report our recent results on the development of a novel strategy for the stereoselective construction of the tetracyclic core of Aspidosperma alkaloids. Our method involves an Ir-catalyzed PET reaction of K for the stereoselective formation of the cis-configured BC bicyclic core with an all-carbon quaternary center [25–26]. Computational studies suggest that the observed tandem PET reaction of K proceeds via an unusual 1,3-C shift to afford M, i.e. through an interrupted [2 + 2] cyclization/retro-Mannich reaction.
Results and Discussion
Conditions optimization
Our study commenced with the evaluation of the proposed PET-based tandem [2 + 2] cyclization/retro-Mannich reaction. We selected compound 10a, which has a spiro-pentacyclic core, as the target to be synthesized via the proposed PET reaction of substrate 9a (Table 1). Substrate 9a can be synthesized from tryptamine and a substituted cyclobutane-1,3-dione according to a published protocol [27].
Initially, we focused on identifying catalysts that could promote the proposed PET reaction of 9a on the basis of our previous results [15]. The reaction was performed in MeCN in the presence of photocatalysts under blue light-emitting diode (LED) irradiation at 30 °C. The results are listed in Table 1. Catalysts I [28], II [29], and III [30] gave the desired product 10a, with catalyst I giving the best result (Table 1, entries 1–3). The necessities of irradiation and the presence of a photocatalyst were also defined (Table 1, entries 4 and 5). However, use of the other tested catalysts did not give the desired product under the reaction conditions (see Supporting Information File 1 for details). Next, we evaluated the effects of different solvents on the production of 10a in the presence of catalyst I. Changing the solvent to MeOH, tetrahydrofuran (THF), and dichloromethane (DCM) resulted in decreased yields and substrate conversions, and intense substrate decomposition (Table 1, entries 6–8). These results showed that MeCN is the best solvent for the reaction.
Finally, we investigated the effects of an alternative N-substituent in the indole moiety, and the reaction time and temperature on the photocyclization results. Boc-substituted substrate 9f was subjected to the optimized conditions, which resulted in both a lower conversion and yield (Table 1, entry 9). In contrast, when the reaction of 9f was performed in a mixed MeCN/toluene 10:1 solvent, the conversion of substrate 9f to product 10f decreased, but the yield increased to 56% (Table 1, entry 10) compared with that in entry 9 (42%). These results indicate that Cbz is a more effective protecting group than Boc under the profiled conditions. The effects of the reaction temperature on the outcome of the PET reaction of 9a were investigated. Among the conditions screened (Table 1, entries 11–13), the reaction in MeCN at 30 °C for 24 h gave the best result, namely a quantitative conversion and 90% yield.
Substrate scope
With the optimal conditions in hand, we then explored the substrate scope. Targeting on the total synthesis of Aspidosperma alkaloids, different tryptamine-substituted cyclobutenones 9a–n were prepared and reacted under the optimal conditions. The results are shown in Scheme 1. Substrate 9a, which contains a spiro-cyclopentene moiety, delivered the best result and gave 10a in 86% isolated yield. When C15 or C16 was substituted with a chlorine atom, 10b and 10c were obtained in 70% and 72% yield, respectively. However, when C15 was substituted with a methyl group, the yield of 10d decreased slightly to 63%. Product 10e, which has two contiguous quaternary stereogenic centers at C2 and C12, was obtained in 74% yield without a significant change in the yield, which indicates that steric hindrance at C2 has a limited effect on the reactivity. However, when the steric hindrance of the protecting group was increased by replacing –Cbz with –Boc, the activity of the PET reaction decreased, which resulted in a lower yield of 10f (45%). In the case of substrate 9g, which has a stereogenic centre at C10, photocyclization afforded 10g in moderate yield and with excellent diastereoselectivity (76% yield, dr >20:1). This indicates that the reaction is controlled by the substrate conformation.
Substrate scope.
To investigate the effects of the spirocyclic ring size on the photocyclization, substrates 9h and 9i were prepared and subjected to the optimal conditions. The PET reaction of 9h with a spiro-cyclohexene unit gave a pair of separable isomers, 10h-1 and 10h-2, in 86% yield and a 1:1 ratio. The reaction of 9i, which bears a spiro-cycloheptene moiety, afforded the annulated product 10i in 67% yield. This indicates that an increase in the spirocyclic ring size negatively affects the photocyclization outcome. We expanded the reaction scope by synthesizing substrates 9j–l, which contain various types of saturated spirocycles. As anticipated, 10j, 10k, and 10l were obtained in moderate yields (57–66%) under the standard reaction conditions. When substrates 9m and 9n, which have a gem-dimethyl and gem-diethyl group, respectively, were used, their PET reactions required a longer reaction time to achieve full conversion. The resulting products 10m and 10n were obtained in yields of 62% and 58%, respectively.
Computational study
The synthesis of (±)-aspidospermidine (1) and (±)-limaspermidine (2) showcased the effectiveness of our strategy for constructing complex monoterpene indole alkaloids [26]. In this work, we turned our attention to investigating the mechanistic intricacies of the key PET reaction for formation of the unique bicyclo[2.2.0]hexane unit present in the proposed intermediate L (Figure 1). In the presence of the excited photocatalyst [FCNIr(III)Pic]*, the substrate participates in an oxidative single-electron transfer (SET) process, which leads to the formation of IN1. The radical cation IN1 served as the reference point for DFT investigations. As illustrated in Figure 2a, facilitated by a favorable radical cation–π interaction [31], IN1 proceeds to the first radical addition transition state (TS1), with an energy barrier of 8.3 kcal/mol. This leads to the formation of benzyl radical IN2, which has a boat-like seven-membered ring. This structural feature may facilitate formation of the C19–C12 bond, even in the presence of critical steric hindrance between the C5 quaternary carbon and the indole moiety.
Computational study. a) Energy profiles from IN1 to IN3 and spin density of TS2 (isovalue = 0.004), solvated (SMD) Gibbs free energies were calculated at the B3LYP-D3/def2-tzvp//B3LYP/6-31G(d) level in MeCN. b) Spin density of IN4 (isovalue = 0.004). c) IRC analysis of TS2 (red line) and bond-length changes of C19–C3 (yellow line) and C19–C12 (blue line) along IRC path. d) Mayer bond order changes of selected structures along IRC path. e) Spin-density analyses of R-24 and F-10 (isovalue = 0.004). f) NBO analyses of the selected transient structures (isovalue = 0.04).
Further DFT investigation proved challenging because of the peculiar features that the potential energy surface (PES) exhibits in the rate-determining step, which involves both formation of the C19–C12 bond and cleavage of the C19–C3 bond. This process has an energy barrier of 21.8 kcal/mol via TS2, and results in direct formation of IN3, which can undergo reductive SET in the presence of [FCNIr(II)Pic]^−^. This leads to the regeneration of [FCNIr(III)Pic] to complete the catalytic cycle and formation of the final product via proton transfer. Intrinsic reaction coordinate (IRC) analysis showed the reaction coordinate connecting the transition state TS2, the reactant (IN2), and the C19–C3 bond cleavage product IN3 wells (Figure 2c).
Possible intermediates with a bicyclo[2.2.0]hexane unit located on the PES minimum were investigated by performing two-dimensional scans of simplified structures to find a minimum. When the nonbonding sp^3^ orbital of the N atom is positioned antiperiplanar to the adjacent C19–C3 bond, an intermediate IN4 with a bicyclo[2.2.0]hexane unit was successfully located. However, the Gibbs free energy of IN4 (ΔG° = +27.4 kcal/mol) is significantly higher than the activation energy of TS2 (ΔG^‡^ = 22.4 kcal/mol). This energy difference can be attributed to a subtle discrepancy between the spin localizations of IN4 and TS2. As depicted in Figure 2b, the spin density of IN4 is predominantly concentrated at the benzene ring, whereas in TS2 it is primarily localized at C19, C12, C3, and N9 (Figure 2a) [32]. Therefore, the intermediacy of IN4 was ruled out.
We assume that the overlap between the sp^2^-hybridized N spin center and σ* (C19–C3) in TS2, and the ring-strain release of the transient bicyclo[2.2.0]hexane unit, play essential roles that enable the reaction to occur. Thus, natural bond orbital (NBO) [33] and Mayer bond order [34–35] analyses were performed to determine the hyperconjugative interactions in the intermediates, transition states, and transient structures along the IRC path (Figure 2c and 2d). In the early stage of this process (Figure 2f, IN2 → R-50 → R-24 → TS2), formation of the C19–C3 bond arises from the orbital overlap between the p orbital of the C19 atom (Figure 2e, R-24) and the π_(C3=N9)* orbital. During this process, the bond order of C19–C12 increases, but no significant change is observed for C19–C3. This leads to the formation of a nonbonding p orbital at the N9 atom (Figure 2e, F-10). Further geometrical adjustment and conformational restriction of the transient structure enable the N9 nonbonding p orbital to align parallel to the σ(C19–C3)_* orbital (Figure 2f, TS2 → F-10 → F-40 → IN3), which reinforces the hyperconjugative interaction. Facilitated by the bond stretching and bond-angle bending of the transient structure with a pseudo bicyclo[2.2.0]hexane unit, the favorable hyperconjugative interaction ultimately leads to cleavage of the C19–C3 bond (TS2 → IN3) and release of the ring strain.
DFT analysis hereby explains that the orbital symmetry involved in this process does not conform to a sigmatropic rearrangement reaction [36]. Inspection of the changes in the bond lengths (Figure 2c) and Mayer bond orders (Figure 2d) along the IRC path clearly show that C19–C12 bond formation and C19–C3 bond cleavage are asynchronous. On the basis of these premises, we assume that the process IN2 → IN3 is neither strictly concerted nor stepwise [37–38]. This can be attributed to the inherent ring-strain release in the transient structure located on the PES and the hyperconjugative interaction between the N9 nonbonding p orbital and σ_(C19–C3)_* during geometrical distortions. This ambiguous mechanistic feature suggests an unusual 1,3-C shift, and indicates that this reaction proceeds via a PET-initiated interrupted [2 + 2]/retro-Mannich process.
Conclusion
In summary, a PET-initiated cationic radical-derived interrupted [2 + 2]/retro-Mannich reaction has been developed for constructing the ABCE tetracyclic cores of Aspidosperma alkaloids from tryptamine-substituted cyclobutenones. Importantly, this methodology has already been successfully applied in the total syntheses of (±)-aspidospermidine and (±)-limaspermidine using 10a and 10h as substrates, respectively [26]. The functionalized C5 atom in the formed ABCE tetracyclic core provides potential opportunities for accessing more complex indole alkaloids. The extensive DFT study indicated that the observed PET-initiated cationic radical-derived reaction proceeds via an unconventional formal 1,3-C shift, which is neither concerted nor stepwise. These findings shed light on the mechanistic innovation of a PET-initiated radical-derived reaction that was driven by the ring-strain release.
Supporting Information
File 1Experimental procedures, characterization data, NMR spectra, and computational study.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bach T Hehn J P Angew Chem, Int Ed 2011501000104510.1002/anie.20100284521246702 · doi ↗ · pubmed ↗
- 2Kärkäs M D Porco J A Jr Stephenson C R J Chem Rev 2016116179683974710.1021/acs.chemrev.5b 0076027120289 PMC 5025835 · doi ↗ · pubmed ↗
- 3Pitre S P Overman L E Chem Rev 20221221717175110.1021/acs.chemrev.1c 0024734232019 · doi ↗ · pubmed ↗
- 4Winkler J D Bowen C M Liotta F Chem Rev 1995952003202010.1021/cr 00038 a 010 · doi ↗
- 5Baldwin J E Thermal Cyclobutane Ring Formation Comprehensive Organic Synthesis 19915 Oxford, UK Pergamon Press 638410.1016/b 978-0-08-052349-1.00120-7 · doi ↗
- 6Poplata S Tröster A Zou Y-Q Bach T Chem Rev 20161169748981510.1021/acs.chemrev.5b 0072327018601 PMC 5025837 · doi ↗ · pubmed ↗
- 7Großkopf J Kratz T Rigotti T Bach T Chem Rev 202212221626165310.1021/acs.chemrev.1c 0027234227803 · doi ↗ · pubmed ↗
- 8Takahashi Y Okitsu O Ando M Miyashi T Tetrahedron Lett 1994353953395610.1016/s 0040-4039(00)76711-6 · doi ↗