On-Surface Synthesis of Azobenzene-Linked Porphyrin Derivatives
Yuji Isshiki, Donglin Li, Saranyan Vijayaraghavan, Kewei Sun, Huynh Thien Ngo, Luiza Buimaga-Iarinca, Yoshitaka Matsushita, Edward A. Neal, Cristian Morari, Jonathan P. Hill, Shigeki Kawai

TL;DR
This study explores how the chirality of porphyrin structures affects on-surface synthesis reactions, revealing that homochiral arrangements lead to lower reaction yields.
Contribution
The novel contribution is demonstrating how molecular chirality in self-assembled structures influences azobenzene formation yields on surfaces.
Findings
Homochiral porphyrin assemblies show lower azobenzene formation yields compared to heterochiral ones.
Disassembly of homochiral structures is necessary for the reaction, reducing efficiency.
Machine learning analysis confirmed statistical differences in reaction outcomes based on chirality.
Abstract
On-surface synthesis has become an attractive strategy to obtain functionalized carbon nanostructures from small precursor molecules using a bottom-up approach. Although various on-surface reactions have been developed, it is still unclear how the chirality of self-assembled structures prior to reaction affects the coupling process. Here, we investigate homocoupling of nitro-phenyl groups in Pt-porphyrin derivatives on Au(111) surfaces using low-temperature scanning tunneling microscopy. Two different self-assembled structures composed either of linear oligomer of molecules of opposing chirality or of discrete trimers of molecules having the same chirality (i.e., homochiral) were respectively obtained by using differently substituted phenyl and 3,5-di-tert-butylphenyl groups porphyrin cores. A machine-learning-assisted protocol was used for large-scale statistical analysis revealing the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1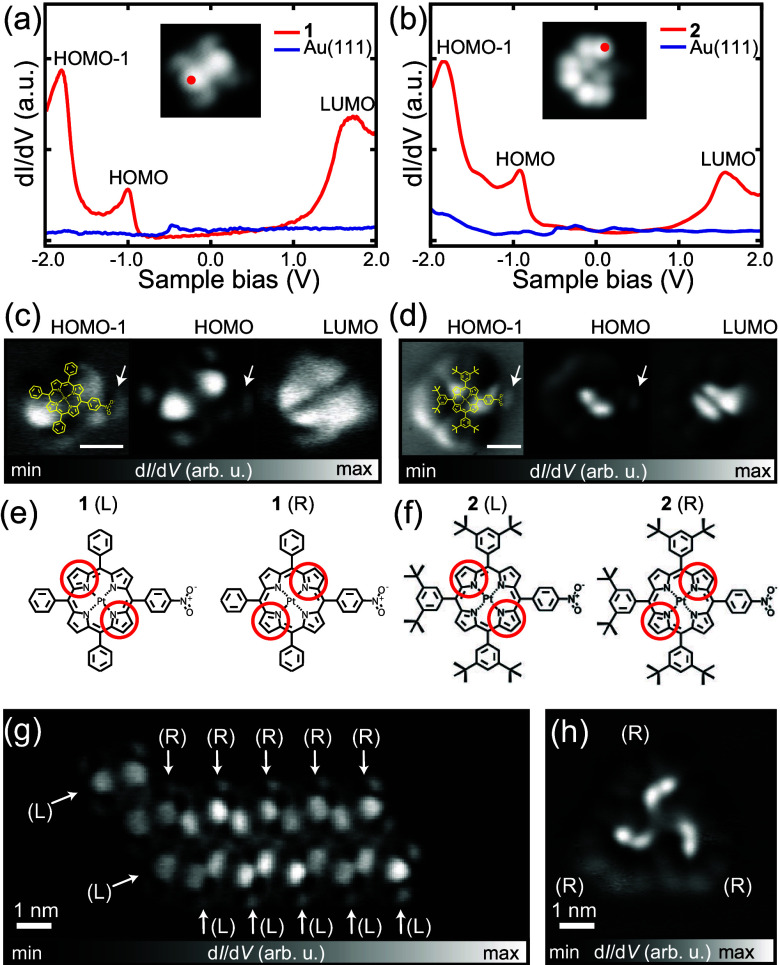 Figure 2
Figure 2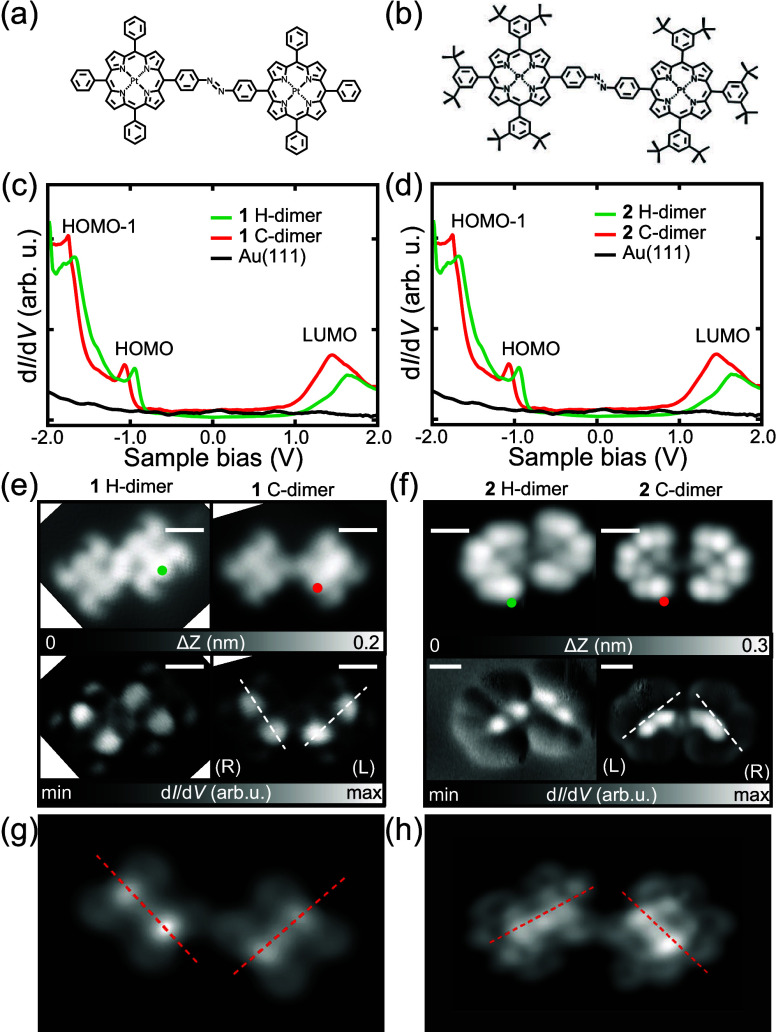 Figure 3
Figure 3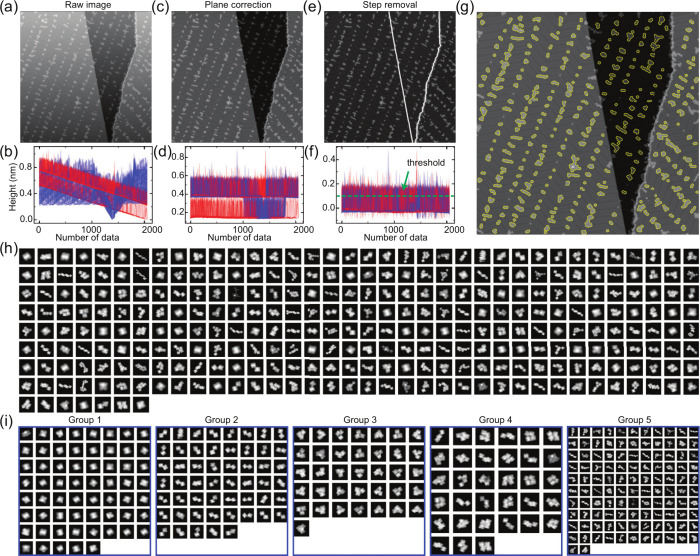 Figure 4
Figure 4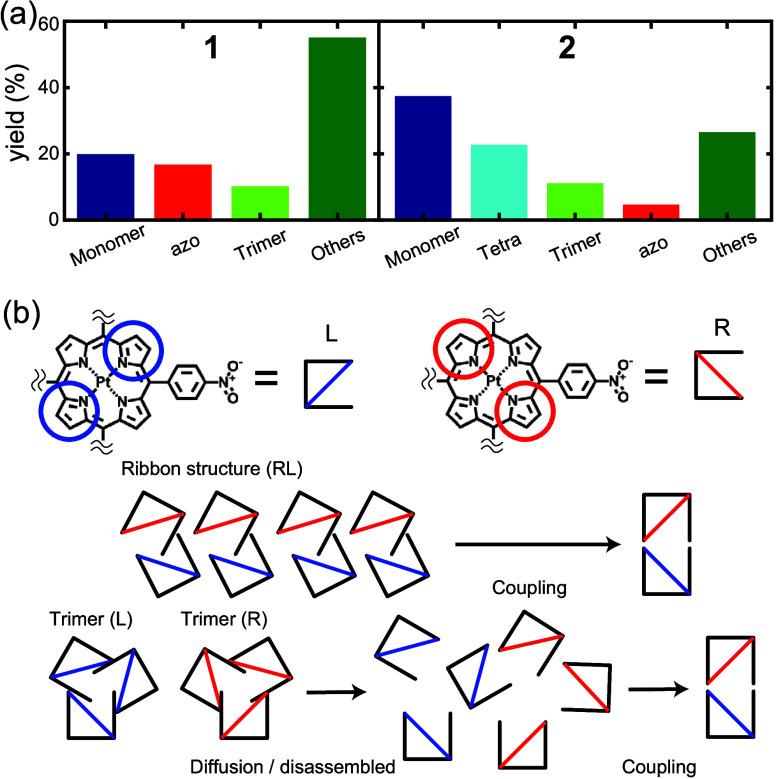 Figure 5
Figure 5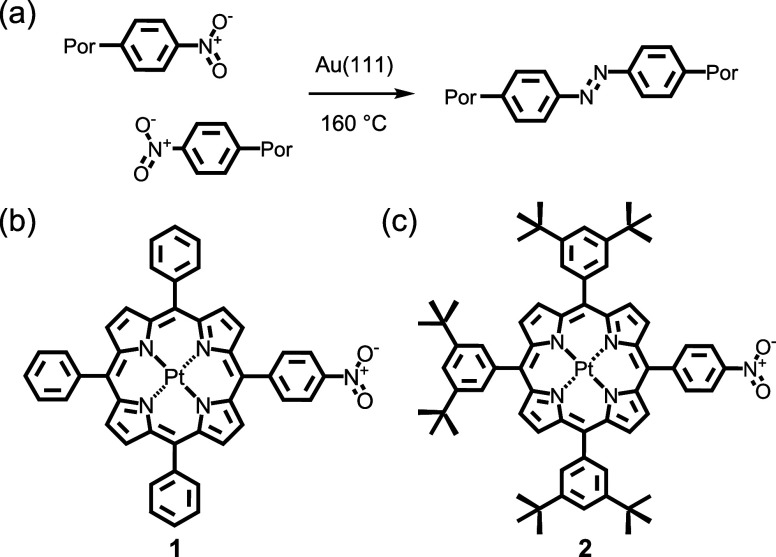 Figure 6
Figure 6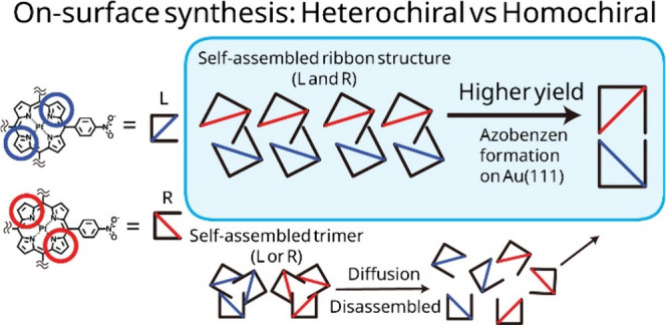 Figure 7
Figure 7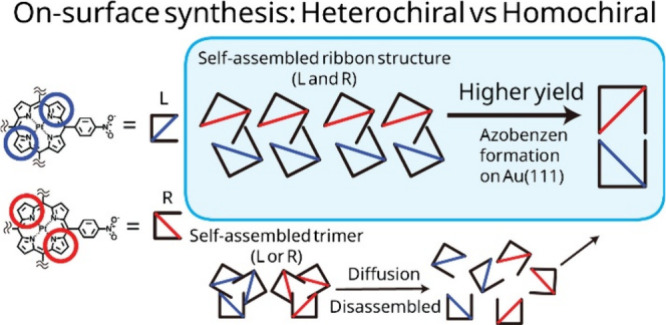 Figure 8
Figure 8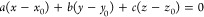 Figure 9
Figure 9- —Ministerul Cercetarii, Inovarii si Digitalizarii10.13039/100018987
- —Japan Society for the Promotion of Science10.13039/501100001691
- —Japan Society for the Promotion of Science10.13039/501100001691
- —Japan Society for the Promotion of Science10.13039/501100001691
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSurface Chemistry and Catalysis · Molecular Junctions and Nanostructures · CO2 Reduction Techniques and Catalysts
Porphyrins play important roles in a wide variety of natural and synthetic systems from biological catalysts to molecular electronic devices. ?−? ? ? Their ability to coordinate metals makes them essential components of catalytic? or light-harvesting systems,? and so on. In particular, π-extended porphyrin molecules are attracting attention in the field of reversible photoisomerization and photoresponsive conjugated molecules. ?−? ? To this end, various bonds between alkenes, alkynes, imines, and azo groups have been formed to extend the conjugated systems. Of these, azo groups have relatively strong electronic exchange with porphyrins? due to orbital matching of porphyrins with azo bridges based on continuous sp? conjugation and fewer steric effects (compared to, e.g., alkenes).
On-surface chemistry is a promising bottom-up approach to molecular systems assembly and synthesis and has, for example, enabled the creation of designer carbon nanostructures through the coupling of small organic molecules adsorbed on surfaces.? In particular, since the first synthesis of graphene nanoribbons (GNR) in 2010,? this field has developed rapidly. Numerous homogeneous reactions ?−? ? have been extensively studied on metal surfaces to construct functional nanostructures using carefully designed precursor molecules. Among the various precursor molecules, porphyrins stand out as particularly attractive building blocks due to their versatile coordination chemistry, which enables tuning of electronic, optical, and magnetic properties. To this end, significant advancements involving porphyrins have been achieved, including the synthesis of π-extended porphyrins, ?,? hierarchical extension in two-dimensions,? porphyrin–GNR hybrids,? block oligomers, ?,? and quantum nanomagnets.? Beyond the construction of functional nanostructures, an understanding of the mechanisms of these reactions is also crucial, as they differ significantly from traditional wet chemistry reaction pathways. Reactions occurring at a surface are profoundly influenced by different surface effects, including catalytically active adatoms,? surface mobility,? and lattice-guided interactions.? While self-assembled structures have been demonstrated to effectively modulate the activity and selectivity of surface reactions, ?,? reactions involving chirality at a surface remain relatively unexplored. It is anticipated that the unique assembly patterns of chiral molecules hold strong potential to steer reaction sites and influence reaction mechanisms.
Here, we present the on-surface homocoupling between nitrobenzene groups, which are connected to the meso-positions of porphyrin derivatives (Scheme). To investigate the influence of the relative position of the reactant, two different molecular assemblies were formed by substituting simple phenyl or 3,5-di-tert-butyl-phenyl groups to the porphyrins. Using scanning tunneling microscopy (STM), we have found that the initial self-assembled structure affects the efficiency of azobenzene formation. This research provides insights into the on-surface synthesis using chiral precursors.
The 4-nitrophenyl-substituted compounds used in this work were selected for their known propensity to undergo reaction to form substituted azobenzene derivatives on Au(111).? Furthermore, platinum(II) complexes of tetraphenylporphyrin derivatives were selected for investigation not only for their excellent stability but also because of the expectation that the porphyrin macrocycle is ‘locked’ in a planar configuration, ?,? although here we show that these compounds exhibit the frequently observed tendency of porphyrin macrocycles to adopt a saddle conformation in response to deposition on a metal substrate.? The structures of the frontier molecular orbitals of the platinum porphyrins also indicate little or no contribution to their structures by the central metal cation (see )
The complex 5-(4-nitrophenyl)-10,15,20-triphenylporphyrin platinum(II) (1) was deposited by sublimation at 533 K onto a clean Au(111) surface maintained at room temperature. After cooling to 4.3 K, the formation of self-assembled ribbon structures was observed (Figurea). The ribbon structures consist largely of double rows (Figureb), aligned with the fcc herringbone structure. Individual molecules appear to have 2-fold symmetry based on a saddle-shaped conformation of the metalloporphyrin macrocycle when adsorbed at a surface. ?,? Molecules in both rows have the same mutual orientation with an angle of 55° subtended between molecular normal axes passing through Pt atom and two opposing pyrrole rings (Figurec). Adjacent Pt core atoms are separated at distances of 1.50 nm (intrarow) and 1.76 nm (inter-row). The preference for double row formation is caused by hydrogen bonding between the nitro group and C–H groups of the porphyrin (Figurec).? Since the single nitro group involved in hydrogen bonding is concealed at the interior of the double row island, there is no two-dimensional extension of the structure unless the molecular coverage reaches one monolayer (). Also, no intermolecular covalent bonding could be detected, and individual molecules could be separated from the molecular assembly by mechanical manipulation using the STM tip ().? At Au(111) elbow sites, hydrogen bonding (H−) dimer and trimer structures of 1 were also observed (). Next, 5-(4-nitrophenyl)-10,15,20-tris(3,5-di-t-butylphenyl)porphyrin platinum(II) (2) was deposited on clean Au(111) maintained at room temperature. Small molecular clusters were observed adsorbed at the elbow sites (Figured). Magnified views of the topography show that the cluster is composed of three molecules in a windmill-like shape (Figuree). The Pt–Pt distance of 1.75 nm is similar to that found in the ribbon structure of 1. A model of the chemical structure (Figuref) indicates that 2 H-trimers are also formed by hydrogen bonds between nitro groups and the porphyrin cores. H-Dimers of 2 were also occasionally observed (Figure S4c). Similar interactions albeit of differing geometry can also be found in the X-ray crystal structure of 2 (). It is likely that the bulky tert-butyl groups prevent the formation of the ribbon-like self-assembly found in 1 since van der Waals interactions between tert-butyl groups are relatively large favoring the H-trimer structure.
In both molecules, the dI/dV spectra of the monomer recorded in a bias range of – 2.0 to 2.0 V contain three distinct peaks at 1.7, – 1.0, and – 1.8 V, which correspond to the lowest unoccupied molecular orbital (LUMO), highest occupied molecular orbital (HOMO), and HOMO–1 levels, respectively (Figurea,b). Thus, the peripheral phenyl and ditert-butyl-phenyl groups do not affect the orbital energies of the porphyrin cores. The dI/dV map of 1 taken at the HOMO energy has two bright regions at the porphyrin core. This is similar to that of 2 but the two bright regions appear in closer proximity due to the larger dimensions of 2 (Figuresc and ?d). LUMO orbitals are also distributed on the porphyrin core similarly for 1 and 2 (Figuresc and ?d, right sides). The nitro groups appear as bright tails as indicated by white arrows, which are more obvious in the HOMO–1 orbitals shown in the left-hand panels of Figuresc and ?d. The two bright spots and the tail establish the prochirality of the molecule on surface as defined by right (R) or left (L) angles relative to the nitro group axis (Figurese and ?f). Note that the dI/dV spectra and maps of 1 ribbon and 2 trimer are almost identical to those of the monomer (). We found that the ribbon is composed of 1 with both chiralities with molecules in the upper row exhibiting R chirality and those in the lower row exhibit L chirality (Figureg). In contrast, the trimer of 2 is composed of molecules with exclusively either R or L chirality (Figuresh). Ribbon and trimer structures are most likely formed through intermolecular hydrogen bonding interactions involving nitro groups. In fact, C–H···O H-bonding is an important interaction in self-assembling and biochemical systems. ?,? For 1, strong N–O···H–C H-bonding occurs between a nitro group of one molecule and an adjacent phenyl ring on the opposing row of molecules of opposite chirality (see also ). For 1 ribbons, strong hydrogen bonding involving nitro groups forces the self-assembled porphyrin units to be incommensurate with the gold substrate. The resulting different adsorption sites between R and l isomers results in variations in the observed contrast in the STM images, which is further enhanced by the intrinsic chirality of the structure. For 2, There is a 3-way mutual interaction between nitro groups and the proton ortho to the nitro group in the adjacent molecules. Based on the dimensions, there is also the possibility of further stabilization of the trimer unit by further interaction with an adjacent t-butyl group (). Based on reported N–O···H–C H-bonding distances, ?,? inter-Pt distances for 1 of 1.8 Å (inter-row) and 1.3 Å (intrarow) are found, which support the model structure. For 2, a Pt–Pt distance of 1.75 Å estimated from the model agrees well with that found experimentally.
Both the ribbon and H-trimer structures are eliminated by annealing at 160 °C for 10 min () followed by the emergence of small molecular clusters (dimers) absorbed mostly at the elbow sites on Au(111). In both cases, the whole dimers of 1 and 2 can be manipulated by using the STM tip () indicating that covalent bonds have been formed between porphyrin cores. Characteristic phenyl and 3,5-ditert-butyl-phenyl groups remain, indicating that the nitro groups have reacted in a reductive homocoupling leading to the synthesis of azobenzene moieties (Figuresa,b) as has been observed in previous work.? The dI/dV spectra of covalent bonding (C−) dimerized 1 and 2 are almost identical to those of the monomers and H-dimers obtained prior to annealing (Figuresc,d). However, there are significant peak shifts to lower energy by approximately 0.2 eV, which is most probably related to differences in the vacuum level.? Furthermore, calculated Bader charges () for the monomers and azobenzene dimers of 1 and 2 indicate that a large positive charge contribution located at the nitro N atoms of the monomer is eliminated by the reductive coupling process to the azobenzene dimer leading to differences in the charge transfer characteristics between the molecules and metal surface in that region (other parts of the molecules exhibit similar charge transfer prior to and following dimerization; see ). It should also be pointed out here that the reaction temperature of 160 °C is not sufficient to cause cyclodehydrogenation between phenyl substituents and the porphyrin core.? Prior to annealing, the nitro group axis in the H-dimer is offset by 0.5 nm (Figurese,f). In contrast, azobenzene formation results in an almost symmetric linear dimer structure. The dI/dV maps of C-dimerized 1 and C-dimerized 2 indicate that both products are composed of one R and one L chiral units each and, interestingly, we found no azobenzene dimers with two of the same chiral units. Also, note that only the trans configuration of the azobenzene group is formed. For computed structures of the monomers and azobenzene dimers, see .
Since the coexisting RL chirality in C-dimerized 1 is the same as that in the ribbon structure, the self-assembled structure should promote C-dimerization based on nitro groups being in proximity in an appropriate geometry for reaction. In contrast, the self-assembled trimer of 2 composed of molecules of a single chirality as the majority of the self-assembled structure (Figured), must be decomposed prior to reaction to a heterochiral C-dimer because its nitro groups are not in an appropriate proximity or geometry for azobenzene formation to occur. Thus, chiral 2 molecules diffuse on the surface until encountering a molecule in appropriate geometry (i.e., with complementary chirality) suitable for reaction to form a heterochiral C-dimer. Based on the different requirements for dimerization, the identity of the self-assembled structures should affect the yield of dimerization. It is notable in both computed structures of the dimerized compounds (Figureg, ?h, and ) that the azobenzene unit has its different moieties (−C_6_H_4_-, -NN-, -C_6_H_4_-) all coplanar similar to the reported X-ray crystal structures of the parent azobenzene compound.? The apparent necessity of this azobenzene geometry in the final dimerized structure enforces heterochirality in the final product since saddle conformations of each component porphyrin unit must be adopted to accommodate the coplanar azobenzene unit based on steric requirements (). This mechanism causes exclusivity of heterochirality for this dimerization reaction and might have connotations for applications of the related compounds in asymmetric synthesis. Furthermore, the azobenzene core adds the aspect of photoisomerization where trans-azobenzene might be converted to cis-azobenzene by applying an appropriate optical stimulus (ultraviolet light). In the present system, the large steric bulk of the porphyrin units prevents access to the cis isomer in the surface-constrained environment. However, smaller substituents of the azobenzene core might permit access to photoisomerization. This matter is currently under investigation in our laboratories.
To conduct a statistical analysis of the yields of different on-surface species, we used the Python program based on previous studies,? where molecules are classified based on the molecular shape obtained from the STM topography. Further, we have developed a program to extract the outline of molecules with the open-source machine learning library, Scikit-learn in Python to implement the classification algorithm. ?,? Automated image processing was conducted to extract the outlines of the molecules. The measured images (particularly the first image collected directly following sample preparation) are tilted due to unintentional thermal drift and scanner creep mainly in the Z direction (Figurea). The blue and red lines in Figureb represent the cross sections of the STM topography in the X (fast scan) and Y (slow scan) directions, respectively. If there is no step in the image, the image can simply be fitted by
where, (a, b, c) are the orthogonal vectors to the plane and (x 0, y 0, z 0) are points that are on the plane.? Consequently, the plane correction can be performed by subtracting the raw image from this calculated plane image. If steps are present in the image, an extended plane correction process is required. First, the image is divided into a 4 × 4 grid. Then, each of the 16 regions is fitted with , and subsequently the entire image is adjusted based on the best fit from each region (Figuresc and ?d). This plane correction is sufficiently effective to correct images containing up to three steps. Since a gold atomic step is approximately 0.2 nm in height (Figured), areas higher than 0.15 nm correspond to upper terraces and molecules adsorbed on the surface. Since the size of the molecules is a few nm, we eliminated these points only on the upper terrace. Subsequently, the height of the upper terrace was adjusted to that of the lower terrace by subtracting the step height from the upper terrace height (Figurese and ?f). Finally, molecular outlines were detected by applying the Depth-First Search algorithm^46^ to the binary image obtained from Figuree with a threshold of 0.1 nm. Each molecule was enclosed in a bounding box that fits its diagonal length, allowing counting of the molecules in the large-scale topography (Figureg). For shape classification, we used the Spectral Clustering method, with Zernike moment coefficients, contour length, and molecular height as feature parameters (Figureh).? The molecules were grouped into five categories: Groups 1, 2, 3, 4, and 5 correspond to monomers, C-dimers, C-trimers, C-tetramers, and other large molecular clusters, respectively (Figurei). Using this approach, we automatically collected 928 and 1056 molecules from 10 and 11 images with scan sizes in the rage of 100 to 200 nm for reactions involving 1 and 2, respectively. It should be noted that this approach can only be used to differentiate molecules/clusters with significantly different dimensions so that, for instance, unreacted (self-assembled) H-dimers cannot be distinguished from reacted azobenzene C-dimers. Also, large surface contaminants of sufficient size become classified as monomers using this method (see ). The overall accuracy of the method is 95.3%.
Figurea shows that the analysis classified the shapes of products into four and five groups for 1 and 2, respectively, after annealing on Au(111) under identical reaction conditions. We found that the reaction yield of azobenzene from 1 (16%) is four times greater than that from 2 (4%). Thus, this analysis indicates that the homocoupling reaction of nitro groups was promoted by the self-assembled 1 ribbon structure, where molecules of one chirality are in close mutual contact with molecules of the other chirality (Figureb). In contrast, the homochiral self-assembled 2 trimer requires disassembly, subsequent monomer diffusion on the surface, then reaction with another molecule of opposing chirality in order to dimerize. These factors (multistep disassembly, requirement to react with opposing enantiomer) lead to a significant reduction in yield for monomer 2, while the yield of monomer 1 might be assisted by proximity of opposing enantiomers in the ribbon structure. In on-surface syntheses, the chirality of the monomer is one of the most important factors since the chirality pattern tends to be maintained before and after the reaction. ?−? ? However, as shown here, if the product is composed of molecules of opposing chirality, the relationship of monomer units and chirality of preformed self-assembled structures can significantly affect the reaction yield. Although the assembly structures might be unstable at the reaction temperature (160 °C), any intermolecular interactions promoting the condensation to chiral products must occur during a relatively short period of time especially for 1 where disassembly is not required for formation of the chiral product. Regardless, this should still lead to preferential heterochiral reaction of 1 (compared to 2) based on the close proximity of the reactants and their appropriate molecular geometries, and the necessity for 2 to disassemble prior to heterochiral azobenzene formation. Also, given that the vertical distances of the nitro groups from the surface and therefore their reactivities for azobenzene formation ought to be comparable (based on the similar distances from the surface: 2.44 Å (for 1) and 2.47 Å (for 2)), the large difference in reaction yield (4-fold for 1 over 2) also indicates a substantial preference for the formation of heterochiral C-dimer for 1 supporting the role of the self-assembly structure in formation of the heterochiral C-dimer. Other factors such as diffusion and collision rates on the surface also probably affect reaction yield.
This study demonstrates that porphyrin chirality significantly influences the azo coupling process on surfaces. In 1, the self-assembled ribbon structure promotes efficient coupling, resulting in higher yields of azobenzene-linked dimers with alternating chirality. The close molecular packing in these ribbons facilitates the reaction, preserving the original chirality. In contrast, 2 predominantly forms hydrogen bonding trimers, requiring dissociation and diffusion for covalent bonding dimer formation, leading to a lower yield. Our findings reveal that controlling molecular chirality and steric factors can optimize surface reactions, offering insights for designing surface-based molecular systems for nanomaterials and electronics.
Methods
STM Experiments
All experiments were performed using a custom built low-temperature ultrahigh vacuum scanning tunneling microscopy, operating under 1 × 10^–10^ mbar at 4.3 K. Clean Au (111) surfaces (MaTeck, purity: 99.999%) were obtained by repeated cycles of Ar^+^ sputtering and subsequent annealing at 450 °C. The molecules were sublimated from a quartz crucible (Kentax GmbH) at 300 °C while the Au (111) substrate was kept at room temperature. STM topographies were obtained in constant current mode. The differential conductance was measured with the digital lock-in amplifier with a modulating voltage of V AC = 10 mV at a frequency of 512 Hz.
Synthesis
5-(4-Nitrophenyl)-10,15,20-triphenylporphyrin and 5-(4-nitrophenyl)-10,15,20-tris(3,5-di-t-butylphenyl)porphyrin were synthesized according to literature methods. ?,?
5-(4-Nitrophenyl)-10,15,20-triphenylporphyrin platinum(II) (1)
Platinum(II) was inserted into 5-(4-nitrophenyl)-10,15,20-triphenylporphyrin (200 mg, 0.3 mmol) exactly according to Turner et al.? Yield: 160 mg (62%). ^1^H NMR (400 MHz, CDCl_3_) δ = 8.75 (d, J = 5.3 Hz, 2H), 8.72 (m, 4H), 8.66 (d, J = 5.3 Hz, 2H), 8.58 (d, J = 8.8 Hz, 2H), 8.34 (d, J = 8.8 Hz, 2H), 8.11 (m, 6H), 7.73 (m, 9H) ppm. ^13^C NMR (101 MHz, CDCl_3_) δ = 148.1, 141.3, 140.9, 140.0, 134.6, 133.7, 131.0, 130.8, 130.6, 129.8, 127.9, 126.9, 122.9, 122.7, 121.9, 119.7 ppm. MALDI-TOF-MS (dithranol; + ve) m/z: found 853.3, calc. for C_44_H_28_N_5_O_2_Pt 853.2 ([M + H]^+^).
5-(4-Nitrophenyl)-10,15,20-tris(3,5-di-t-butylphenyl)porphyrin
platinum(II) (2)
5-(4-Nitrophenyl)-10,15,20-tris(3,5-di-t-butylphenyl)porphyrin (0.5 g, 0.5 mmol) was treated with PtCl_2_ (1.5 equiv, 200 mg) in refluxing benzonitrile (50 mL) for 18 h under a dry nitrogen atmosphere. The reaction was performed in two stages: (1) heating for 3h at 110 °C then (2) refluxing for 18 h. After reaction completion, benzonitrile was removed under reduced pressure. The resulting orange solid was subjected to column chromatography (SiO_2_; dichloromethane/hexane 60:40 v/v). Product containing fractions were combined and the solvents were removed under reduced pressure. The resulting bright orange solid was recrystallized from dichloromethane/methanol yielding 2 as dark orange microcrystalline powder. Yield: 405 mg (68%). ^1^H NMR (400 MHz, CDCl_3_) δ = 8.85 (d, J = 5.3 Hz, 2H), 8.84 (s, 4H), 8.65 (d, J = 5.3 Hz, 2H), 8.63 (d, J = 8.8 Hz, 2H), 8.37 (d, J = 8.8 Hz, 2H), 8.03 (d, J = 1.8 Hz, 4H), 8.02 (d, J = 1.8 Hz, 2H), 7.80 (t, J = 1.8 Hz, 16H), 1.52 (s, 54H) ppm. ^13^C NMR (101 MHz, CDCl_3_) δ = 149.0, 148.8, 147.8, 141.5, 141.3, 141.1, 140.4, 139.6, 134.6, 131.7, 131.3, 131.1, 129.5, 129.1, 129.0, 124.4, 124.1, 122.0, 121.3, 118.8, 35.1, 31.8 ppm. FTIR (ATR): ν = 2957 (s, C–H (Ar)), 2903 (m, C–H (CH_3_)), 2867 (m, C–H (CH_3_)), 1593 (s, C–C (Ar)), 1523 (s, N–O (NO_2_)), 1342 (s, N–O (NO_2_)), 827 (s, C–H (Ar, p-disubstituted)) cm^–1^: HRMS (ESI+) m/z: found 1189.5647, calc. for C_68_H_76_N_5_O_2_Pt 1189.5641 ([M + H]^+^).
X-ray Crystallography
Crystals suitable for X-ray diffraction were grown by slow diffusion of hexane into a solution of 2 in dichloromethane. Data collection was performed using MoKα radiation (λ = 0.71073 Å) on a RIGAKU VariMax Saturn diffractometer equipped with a charge-coupled device (CCD) detector or a Bruker APEX CCD diffractometer. Prior to the diffraction experiment the crystals were flash-cooled to the given temperature in a stream of cold nitrogen gas. Cell refinements and data reductions were carried out using the d*trek program package in the CrystalClear software suite.? The structures were solved using a dual-space algorithm method (SHELXT)? and refined by full-matrix least-squares on F2 using SHELXL-2014? in the WinGX program package.? Non-hydrogen atoms were anisotropically refined and hydrogen atoms were placed on calculated positions with temperature factors fixed at 1.2 times Ueq of the parent atoms and 1.5 times Ueq for methyl groups. Crystallographic data (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre with CCDC reference numbers 2350301 (2). Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK http://www.ccdc.cam.ac.uk, e-mail: [email protected], or fax: + 44 1223 336033.
Computational Methods
The simulations were performed using the Siesta code. ?,? As exchange-correlation functional, we have used the GGA-PBE functional.? The DFT interactions were supplemented with a set of Grimme force-fields, to simulate the dispersion interactions (van der Waals).? The systems were confined to unit cells that allow the study of periodically Au(111) surface and included 3 atomic layers for all the investigated systems. The supercell sizes vary from 10 × 10 Au atoms for monomer s adsorption to 9 × 15 Au atoms for C-dimers. The length of the cells along the OZ axis was 40 Å for all systems, thus allowing a vacuum level of more than 25 Å that we consider large enough to avoid the artificial influence of the electric charge from one cell to another. The Monkhorst–Pack grid for the integrals in the Brillouin zone was 1 × 1 ×
- As relaxation methods, we used the Conjugated Gradient for all the systems. As basis sets, we used double-ζ polarized (DZP) with an energy shift of 100 meV for all atoms. The systems were allowed to find the local minima until the maximum gradient in the relaxed structure was below 0.04 eV/A. The simulations of STM images were performed by using the Tersoff-Hamman approximation. That method considers that the STM image is determined by the local density of states (LDOS).? We computed LDOS in the energy window spanning from the Fermi level of the system up to 1 eV below the Fermi level, corresponding to an external bias of 1 V. The surface was scanned by searching for a given constant value of the LDOS (essentially a ″constant current″ STM experiment).
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Marbach H.Surface-Mediated in situ Metalation of Porphyrins at the Solid–Vacuum Interface Acc. Chem. Res.2015482649265810.1021/acs.accounts.5b 0024326308682 · doi ↗ · pubmed ↗
- 2Park J. M.Hong K.-I.Lee H.Jang W.-D.Bioinspired Applications of Porphyrin Derivatives Acc. Chem. Res.2021542249226010.1021/acs.accounts.1c 0011433891405 · doi ↗ · pubmed ↗
- 3Jurow M.Schuckman A. E.Batteas J. D.Drain C. M.Porphyrins as Molecular Electronic Components of Functional Devices Coord. Chem. Rev.20102542297231010.1016/j.ccr.2010.05.01420936084 PMC 2950646 · doi ↗ · pubmed ↗
- 4Robertson N.Mc Gowan C. A.A Comparison of Potential Molecular Wires as Components for Molecular Electronics Chem. Soc. Rev.2003329610310.1039/b 206919 a 12683106 · doi ↗ · pubmed ↗
- 5Jing J.Yang J.Zhang Z.Zhu Y.Supramolecular Zinc Porphyrin Photocatalyst with Strong Reduction Ability and Robust Built-In Electric Field for Highly Efficient Hydrogen Production Adv. Energy Mater.202111210139210.1002/aenm.202101392 · doi ↗
- 6Otsuki J.Supramolecular Approach Towards Light-Harvesting Materials Based on Porphyrins and Chlorophylls J. Mater. Chem. A 201866710675310.1039/C 7TA 11274 B · doi ↗
- 7Esdaile L. J.Jensen P.Mc Murtrie J. C.Arnold D. P.Azoporphyrin: The Porphyrin Analogue of Azobenzene Angew. Chem., Int. Ed.2007462090209310.1002/anie.20060465817278161 · doi ↗ · pubmed ↗
- 8Myles A. J.Branda N. R.Controlling Photoinduced Electron Transfer within a Hydrogen-Bonded Porphyrin–Phenoxynaphthacenequinone Photochromic System J. Am. Chem. Soc.200112317717810.1021/ja 002733 p 11273616 · doi ↗ · pubmed ↗