Effect of the naphthylene linker on the J‐aggregation abilities of chlorophyll‐a derivatives
Yuma Hisahara, Takeo Nakano, Hitoshi Tamiaki

TL;DR
Researchers found that adding a naphthylene linker to chlorophyll-a derivatives enhances their ability to form J-aggregates with unique optical properties.
Contribution
The study introduces a new linker design that improves J-aggregation in chlorophyll-a derivatives, enabling larger red-shifts in their optical properties.
Findings
Chl-a derivatives with a 2,6-naphthylene linker showed a larger red-shift (1270 cm−1) in Qy band compared to p-phenylene derivatives (970 cm−1).
The naphthylene linker promotes single J-aggregation species in micellar solutions, influencing exciton splitting energies.
Computational studies confirmed that linker structure controls chlorin ring positioning in aggregates.
Abstract
Chlorophyll(Chl)‐a derivatives inserting an ethynylene‐naphthylene linker between the chlorin π‐skeleton and hydroxymethyl group were prepared as models of chlorosomal Chls. Their syntheses were achieved via Sonogashira coupling reaction. Their J‐aggregation behaviors were investigated by electronic absorption and circular dichroism spectroscopic measurements. These studies revealed that the 2,6‐naphthylene inserted Chl‐a derivatives gave the single J‐aggregation species in an aqueous Triton X‐100 micellar solution with a larger red‐shift value (1270 cm−1) of the Qy band in spite of its longer linker compared with p‐phenylene inserted Chl‐a derivative (970 cm−1). These unique optical properties were also discussed based on the computational studies, which indicated the different positional relation of chlorin rings in the assemblies by the linker structure. Chlorophyll(Chl)‐a…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
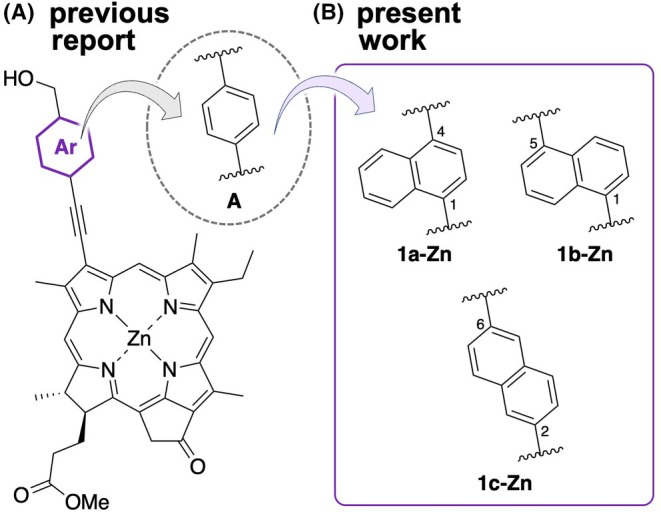 FIGURE 1
FIGURE 1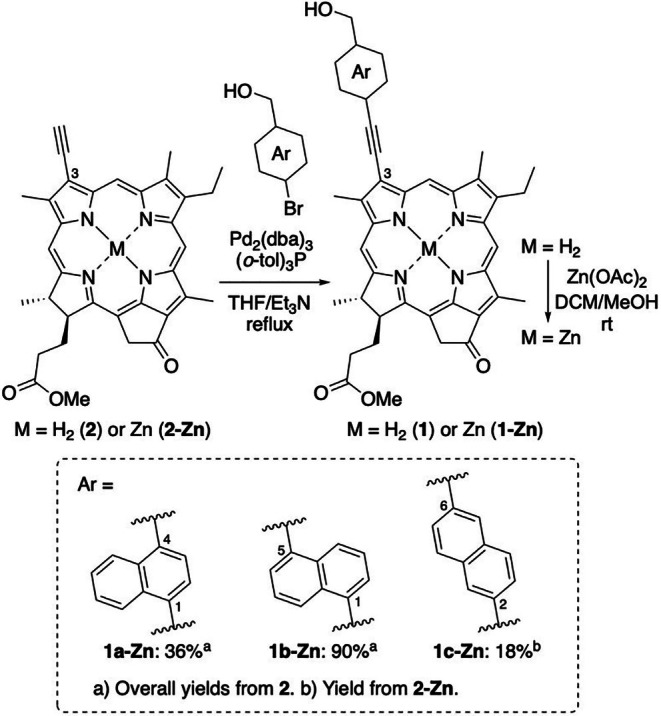 SCHEME 1
SCHEME 1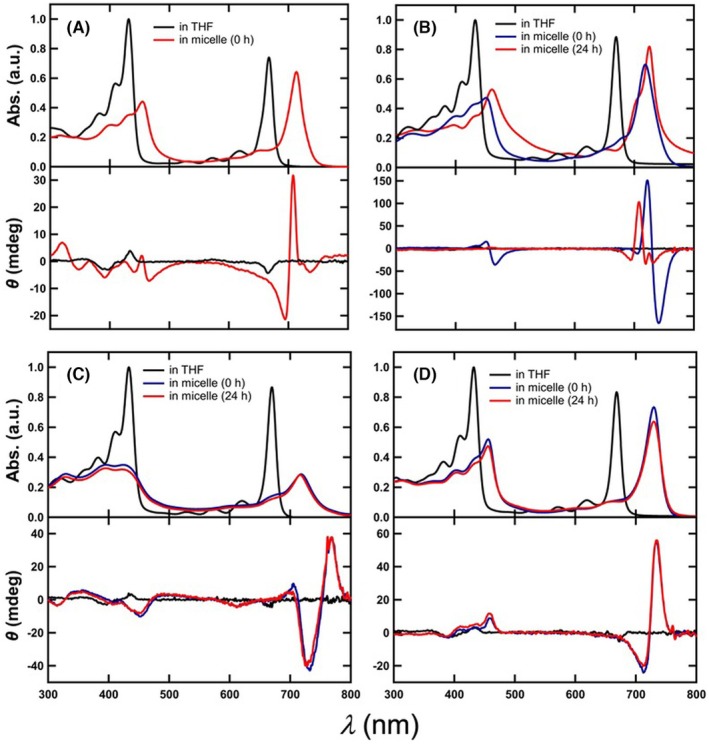 FIGURE 2
FIGURE 2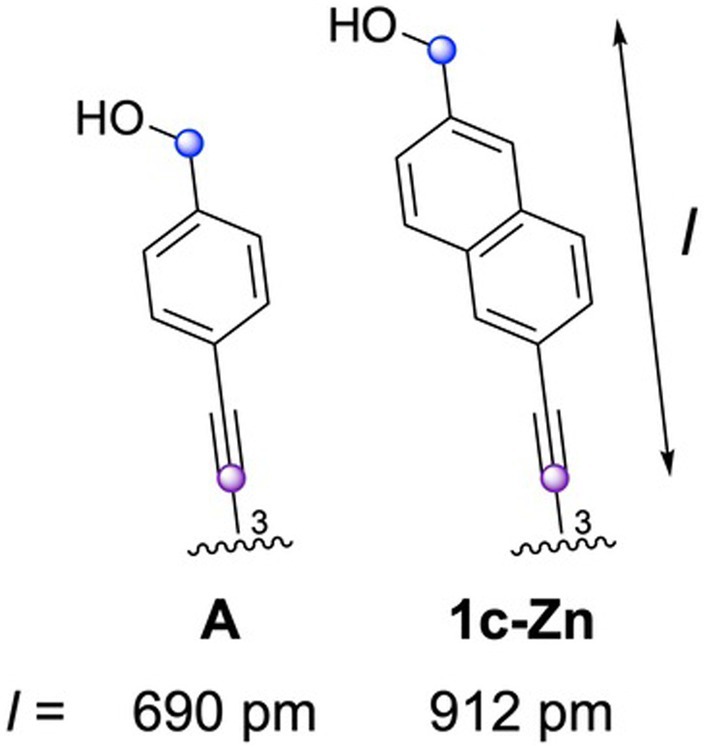 FIGURE 3
FIGURE 3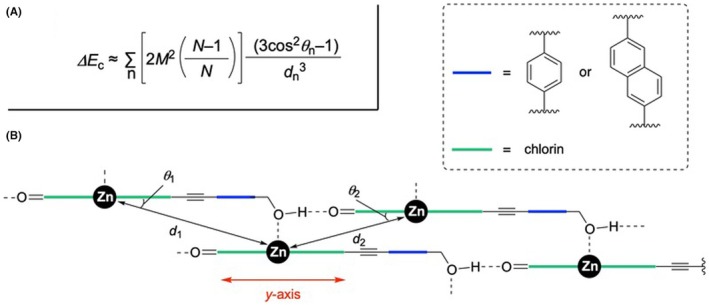 FIGURE 4
FIGURE 4| Chl |
|
| ||
|---|---|---|---|---|
| Soret | Qy | Soret | Qy | |
|
| 431 | 667 | 455 | 713 |
|
| 433 | 669 | 461 | 725 |
|
| 434 | 670 | 424 | 718 |
|
| 432 | 668 | 456 | 730 |
| Chl |
| Δ | Δ | |
|---|---|---|---|---|
| Monomer | Aggregate | |||
|
| 667 | 713 | 970 | 1.92 |
|
| 668 | 730 | 1270 | 2.53 |
- —JSPS KAKENHI
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPhotosynthetic Processes and Mechanisms · Spectroscopy and Quantum Chemical Studies · Porphyrin and Phthalocyanine Chemistry
INTRODUCTION
Supramolecules are generally formed by electrostatic interaction, dipolar interaction, hydrogen bonding, and hydrophobic interaction. Their shapes depend in a large part on the structures of their building blocks, and they exhibit drastically different physical, electronical, and optical properties compared to those of monomers. Therefore, various investigation of supramolecules toward application in material chemistry, such as host‐guest systems,1, 2 self‐healing materials,3, 4, 5 redox catalysts,6 nano‐machines,7, 8 and drug‐delivery systems.9, 10 The supramolecular features are also important in the natural world. For instance, J‐aggregates of bacteriochlorophyll (BChl) molecules act as light‐harvesting antenna, which are essential for photosynthesis.11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37 This antenna consisting of BChl‐c, BChl‐d, BChl‐e, and BChl‐f is called as “chlorosome”,38, 39, 40, 41, 42, 43 and the exploration of chlorosomal antenna based on the synthetic chlorophyll (Chl) molecules are required to realize artificial photosynthesis.
Naturally occurring BChl molecules give their J‐aggregates through hydrogen bond (3^1^‐OH and 13‐C=O) and coordination bond (3^1^‐O and Mg).44, 45, 46 Therefore, synthesis of Chl molecules bearing the similar functional groups could afford the novel building blocks for the chlorosomal photosynthetic antennae. Actually, various Chl molecules bearing various functional groups at their peripheral positions, such as C3^1^‐, C7‐, C8‐, C13^2^‐, C17‐, and C20‐positions,47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62 were synthesized based on zinc 3^1^‐hydroxy‐13^1^‐oxo‐chlorin63, 64, 65 to investigate their J‐aggregation abilities. Further, Chl‐a derivatives inserting an ethynylene and phenylene linker between the hydroxymethyl group and chlorin π‐skeleton were also prepared to control the exciton splitting energies based on a chlorin‐to‐chlorin distance in the resulting self‐aggregates (Figure 1A).66, 67, 68, 69 These studies revealed that the position of the hydroxymethyl group and substituents on the phenylene group were influence on their J‐aggregation behaviors. Thus, the structure of the linker could be fundamental for the control of J‐aggregation abilities of such Chl‐a derivatives and optical properties of the resulting self‐aggregates, which is essential for development of the artificial light‐harvesting antenna. Especially, the previous reports exhibited that the linear linkers, including an ethynylene‐p‐phenylene moiety, gave Chl‐a derivatives good J‐aggregation abilities.66 Based on these studies, we herein designed and synthesized the Chl‐a derivatives inserting a naphthylene linker as the π‐expanded skeleton with a linear‐like structure; we focused the 1,4‐, 1,5‐, and 2,6‐naphthylene linker in which the two substituted C‐C bonds on the naphthalene ring are in parallel (Figure 1B). Their J‐aggregation behaviors were investigated by electronic absorption and circular dichroism (CD) spectroscopy in an aqueous micellar solution.
Ethynylene‐p‐phenylene linker inserted Chl‐a derivative A (A) and ethynylene‐naphthylene linker inserted Chl‐a derivatives 1a‐Zn, 1b‐Zn, and 1c‐Zn in the present work.
RESULTS AND DISCUSSION
Synthesis of Chl‐a derivatives 1‐Zn
Naphthylene and ethynylene linker inserted Chl‐a derivatives 1‐Zn were prepared from C3‐ethynylated Chl‐a derivatives 2 or 2‐Zn via Sonogashira coupling reaction as shown in Scheme 1.70, 71, 72 The free base of Chl‐a derivative 1a inserting a 1,4‐naphthylene linker was synthesized via Pd‐catalyzed Sonogashira reaction of 2 with 1‐bromo‐4‐(hydroxymethyl)naphthalene. After that, regular zinc metalation70 furnished zinc complex 1a‐Zn. In a similar way of 2 → 1a → 1a‐Zn, the Chl‐a derivative 1b‐Zn inserting a 1,5‐naphthylene linker was synthesized from 2 and 1‐bromo‐5‐(hydroxymethyl)naphthalene. Although the Chl‐a derivative 1c was formed via Sonogashira coupling of 2 with 2‐bromo‐6‐(hydroxymethyl)naphthalene, its isolation was difficult due to the inseparable impurities. Therefore, 1c‐Zn was synthesized by the coupling of 2‐Zn with 2‐bromo‐6‐(hydroxymethyl)naphthalene after the zinc metalation of 2. The synthetic details with their spectral data were described in the Materials and method section of Supporting Information.
Synthesis of Chl‐a derivatives bearing a (hydroxymethylated naphthyl)ethynyl group at the C3 position: dba, dibenzylideneacetone; DCM, dichloromethane; THF, tetrahydrofuran; tol, tolyl.
Self‐aggregation of Chl‐a derivatives in an aqueous micellar solution
In our previous report, the electronic absorption and CD spectra of p‐phenylene inserted Chl‐a derivative A were measured (Figure 2A).66 The electronic absorption spectrum of A in tetrahydrofuran (THF) exhibited large and sharp bands at 431 and 667 nm (Table 1), which are called Soret and Qy bands, respectively (Figure 2A, upper, black line). The spectrum was characterized as monomeric A with single axial THF coordinated to the central Zn atom. Meanwhile, a freshly prepared aqueous Triton X‐100 (TX‐100) micellar solution of A exhibited red‐shifted Soret and Qy bands relative to those in THF; λ ^Soret^/λ ^Qy^ = 455/713 nm (Figure 2A, upper, red line). Strong CD signals were observed at the red‐shifted Qy region (Figure 2A, lower, red line). These results indicated chlorosomal J‐aggregation of A in the aqueous micellar solution, based on the previous data observed for its related compounds.73, 74, 75
Electronic absorption (upper) and CD spectra (lower) of A (A), 1a‐Zn (B), 1b‐Zn (C), and 1c‐Zn (D) in THF and an aqueous solution of 0.025% (wt/v) TX‐100 and 1% (v/v) THF.
In the same manner, the self‐aggregation behaviors of the naphthylene‐inserted Chl‐a derivatives 1‐Zn were investigated (Figure 2B–D). Monomeric 1a‐Zn in THF exhibited similar but slightly red‐shifted Soret (433 nm) and Qy bands (669 nm) (Figure 2B, upper, black line) in comparison with those of A. These shifts were ascribed to the expanded π‐conjugated system by inserting a 1,4‐naphthylene group at the C3^2^‐position. An aqueous micellar solution of 1a‐Zn exhibited a red‐shifted Qy band at 719 nm just after preparation of the aqueous solution (Figure 2B, upper, blue line). This behavior was similar to that of A in an aqueous micellar solution; however, the Qy band of 1a‐Zn was changed over time. Namely, the Qy band at 719 nm decreased, and concomitantly the broadened band at 725 nm accompanying a shoulder at 705 nm increased (Figure 2B, upper, red line). Although the reason for this change is unclear, the 1,4‐naphthylene group could affect the stability of the self‐assembly, gradually changing from kinetically controlled (719‐nm absorbing) to thermodynamically controlled (725‐nm absorbing) self‐aggregates. Actually, 1a‐Zn in an aqueous micellar solution gave the similar electronic absorption spectrum after standing for 90 min at 55°C.* However, the broadened absorption bands could indicate the disordered self‐assemblies including various species. The presence of strong bisignate CD signals in the Qy region just after preparation of the aqueous micellar solution was indicative of the J‐aggregation of 1a‐Zn (Figure 2B, lower, blue line). On the other hand, the CD signals in the Qy region after standing for 24 h drastically decreased (Figure 2B, lower, red line); a positive signal was observed around 705 nm, in which the shoulder band appeared in the absorption spectrum. These changes of the CD signals were consistent with the formation of the different self‐assemblies, which were exhibited by the absorption spectra.
The 1,5‐naphthylene inserted Chl‐a derivative 1b‐Zn also did not exhibit the well‐ordered J‐aggregation. Comparing its electronic absorption spectrum in THF (Figure 2C, upper, black line) with that in aqueous micellar solution (Figure 2C, upper, red line), the bathochromic shift was observed; λ ^Qy^ = 670 (in THF) < 718 nm (in micelle). The absorption bands in an aqueous micellar solution were significantly broadened, where red‐shifted bands were overlapped with the corresponding monomeric bands. These results suggested that some disordered self‐assemblies were generated,76 and the monomeric 1b‐Zn had remained. The aggregated 1b‐Zn showed the S‐shaped CD bands at the shifted regions (Figure 2C, lower, red line).
The J‐aggregation behavior of Chl‐a derivative 1c‐Zn was also investigated (Figure 2D). In THF, monomeric 1c‐Zn exhibited slightly red‐shifted Soret and Qy bands relative to those of A (Figure 2D, upper, black line); λ ^Soret^ = 432 (1c‐Zn) > 431 nm (A) and λ ^Qy^ = 668 (1c‐Zn) > 667 nm (A). These bathochromic shifts were due to the π‐expanded structure mentioned above. When 1c‐Zn was dissolved in an aqueous TX‐100 micellar solution, largely red‐shifted Soret and Qy bands appeared in comparison with those in THF. In the Qy region, a remarkably shifted band was observed at 730 nm, and the spectrum hardly changed during standing for 24 h except the slight suppression of the 730‐nm intensity (Figure 2D, upper, blue to red line). This Qy band in an aqueous micellar solution was sharper than those of 1a/b‐Zn and its full width at half maximum (564 cm^−1^) was comparable to that of A (550 cm^−1^), which suggested that single self‐assembly species of 1c‐Zn was favorable in the aqueous micelle. A strong CD couplet at the red‐shifted Qy region indicated that the chlorosomal J‐aggregation of 1c‐Zn occurred (Figure 2D, lower, red line), similarly as in A.
Calculation studies of the exciton splitting energies in self‐aggregates of Chl‐a derivatives
As shown in Figure 2, 1A‐Zn and 1b‐Zn gave the relatively disordered chlorosomal self‐aggregates in an aqueous micellar solution in comparison with A and 1c‐Zn. These results could indicate that a hydroxymethyl group at the α‐position of a naphthalene ring linker put at a disadvantage to form the self‐aggregates. Therefore, exciton splitting energies (ΔE) of A and 1c‐Zn, which gave their well‐ordered chlorosomal self‐aggregates in an aqueous micellar solution, were estimated by their red‐shift values (Δλ) as shown in Table 2.† In our previous report,66 ΔE value of A was decreased in comparison with 3‐hydroxymethylated Chl‐a derivative due to the extended chlorin‐to‐chlorin distance in the expected aggregates by the inserted ethynylene‐p‐phenylene linker. However, ΔE value of 1c‐Zn inserting a longer linker was larger than that of A; ΔE = 2.53 × 10^−20^ (1c‐Zn) > 1.92 × 10^−20^ J (A), namely, the ratio of ΔE(1c‐Zn) over ΔE(A) was 1.32. Actually, the calculation exhibited that the linker length of 1c‐Zn was longer than that of A as shown in Figure 3; l = 912 (1c‐Zn) > 690 pm (A). These results suggested that the linker length is not necessarily a dominant factor to determine the ΔE value in the aggregates. For the further investigation, the calculation studies were carried out based on the expected chlorosomal self‐aggregate model of A and 1c‐Zn (Figure 4). The relationship of the exciton splitting energy with the chlorin‐to‐chlorin distance d and angle θ was expressed as shown in Figure 4A; M represents the magnitude of the transition moment of the chlorin molecules, and N is the molecular number in their self‐aggregates.77 Regarding A and 1c‐Zn, their main chlorin π‐skeletons were identical and their structural difference is the inserted phenylene/naphthylene linker between the hydroxymethyl and C3‐ethynyl group. Therefore, the values of M along the y‐axis (C3–C13 line) of A and 1c‐Zn monomers were assumed to be comparable. The chlorin numbers N in A and 1c‐Zn self‐aggregates were investigated based on the dynamic light scattering (DLS) studies.‡ These results exhibited that the self‐aggregate of 1c‐Zn was larger than that of A, namely the chlorin number N of 1c‐Zn aggregate was also larger that of A aggregate. However, these differences could have little effect on the value of the N − 1/N (≈1) due to the larger values for Ns (N >> 1), which involve in the equation as shown in Figure 4A. These assumptions indicate that the calculated ΔE is dependent in (3cos^2^ θ − 1)/d ^3^ = K.
Estimated lengths (l) of linkers by MM+/PM3 calculations between the C31 and hydroxymethyl carbon atoms of A and 1c‐Zn.
Distances (d) and angles (θ) between chlorin rings in J‐aggregates of A and 1c‐Zn.
The chlorosomal model derived from linker‐inserted Chl‐a derivative A and 1c‐Zn were illustrated in Figure 4B, and their K‐values were determined as the sum of K 1 and K 2, which were estimated from d 1 and θ 1, and d 2 and θ 2, respectively. Our previous report exhibited that K for A was 1.91 × 10^27^ m^−3^.66 In a similar manner, K for 1c‐Zn was determined by d and θ, which were estimated from the energy‐minimalized aggregate model by MM+ calculation; d 1 = 1377 pm, θ 1 = 25.6°, d 2 = 881 pm, and θ 2 = 31.5°. Based on these data, K 1 and K 2 values were calculated to be 5.51 × 10^26^ and 1.73 × 10^27^ m^−3^, respectively. Therefore, K for 1c‐Zn was 2.23 × 10^27^ m^−3^. The ratio of K(1c‐Zn) over K(A) was 1.17, which was a little different from the aforementioned ratio ΔE(1c‐Zn)/ΔE(A) = 1.32, however, the order was identical. These results suggested that the model calculations were useful. Regarding the chlorin‐to‐chlorin distance, d(1c‐Zn) was larger than d(A) due to the longer naphthylene linker; d 1 = 1377 (1c‐Zn) > 1231 (A) pm and d 2 = 881 (1c‐Zn) > 838 (A) pm.§ These larger d could give a smaller K in 1c‐Zn. By contrast, θ(1c‐Zn) was sufficiently smaller than θ(A); θ 1 = 25.6° (1c‐Zn) < 33.1° (A) and θ 2 = 31.5° (1c‐Zn) < 39.7° (A). These smaller angles produced larger cosθ‐values, which resulted in larger K. These two conflicting factors led to ΔE(1c‐Zn)/ΔE(A) > 1, suggesting that the linker structure could drastically affect the chlorin‐to‐chlorin angles, which is one of the dominant parameters to control the excitonic interaction.
CONCLUSIONS
We synthesized Chl‐a derivatives 1a‐Zn, 1b‐Zn, and 1c‐Zn inserting an ethynylene‐naphthylene linker between the hydroxymethyl group and chlorin ring. These syntheses have been achieved via Pd‐catalyzed cross‐coupling reaction of a C3‐ethynylated Chl‐a derivative with the corresponding naphthyl bromides. Their self‐aggregation behaviors were investigated by measurements of electronic absorption and CD spectra in aqueous TX‐100 micelles. Although 1a‐Zn and 1b‐Zn inserting 1,4‐ and 1,5‐naphthylene linkers, respectively, exhibited their reduced self‐aggregation abilities, 1c‐Zn inserting a 2,6‐naphthylene linker gave the well‐ordered self‐aggregates. Interestingly, the larger red‐shift value of the aggregated Qy band in 1c‐Zn in comparison with that in A inserting a smaller p‐phenylene group was observed in spite of the longer linker. Namely, the excitonic interaction between the chlorin π‐skeletons in the self‐aggregates of 1c‐Zn was larger than that of A, while the chlorin‐to‐chlorin distance in the self‐aggregates was extended. The calculation studies based on the chlorosomal self‐aggregate model of 1c‐Zn exhibited the smaller chlorin‐to‐chlorin angles in comparison with those of A, which could enhance the excitonic interaction. These results suggested that the linker structures could drastically change the positional relationship of the chlorin rings in the self‐aggregates, which could be useful to control the exciton splitting energies.
AUTHOR CONTRIBUTIONS
Yuma Hisahara: Investigation. Takeo Nakano: Project administration, Writing – original draft. Hitoshi Tamiaki: Project administration, Supervision, Funding acquisition, Writing – review and editing.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
Supporting information
Data S1.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Pluth MD , Raymond KN . Reversible guest exchange mechanisms in supramolecular host–guest assemblies. Chem Soc Rev. 2007;36:161‐171.17264920 10.1039/b 603168 b · doi ↗ · pubmed ↗
- 2Schneider HJ , Yatsimirsky AK . Selectivity in supramolecular host–guest complexes. Chem Soc Rev. 2008;37:263‐277.18197343 10.1039/b 612543 n · doi ↗ · pubmed ↗
- 3Gemert GML , Peeters JW , Söntjens SHM , Janssen HM , Bosman AW . Self‐healing supramolecular polymers in action. Macromol Chem Phys. 2012;213:234‐242.
- 4Herbst F , Döhler D , Michael P , Binder WH . Self‐healing polymers via supramolecular forces. Macromol Rapid Commun. 2013;34:203‐220.23315930 10.1002/marc.201200675 · doi ↗ · pubmed ↗
- 5Yang Y , Urban MW . Self‐healing of polymers via supramolecular chemistry. Adv Mater Interfaces. 2018;5:1800384.
- 6Ham R , Nielsen CJ , Pullen S , Reek JNH . Supramolecular coordination cages for artificial photosynthesis and synthetic photocatalysis. Chem Rev. 2023;123:5225‐5261.36662702 10.1021/acs.chemrev.2c 00759 PMC 10176487 · doi ↗ · pubmed ↗
- 7Ortiz‐Rivera I , Mathesh M , Wilson DA . A supramolecular approach to nanoscale motion: polymersome‐based self‐propelled nanomotors. Acc Chem Res. 2018;51:1891‐1900.30179450 10.1021/acs.accounts.8b 00199 PMC 6150652 · doi ↗ · pubmed ↗
- 8Ellis E , Mooethy S , Chio WK , Lee TC . Artificial molecular and nanostructures for advanced nanomachinery. Chem Commun. 2018;54:4075‐4090.10.1039/c 7cc 09133 h 29484317 · doi ↗ · pubmed ↗