Restructuring Antiviral Quinazolinone Frameworks to Derive and Optimize Inhibitors of Chikungunya Virus
Caroline M. Roach, Zachary J. Streblow, Yuting Zhang, Tyler J. Ogorek, Alejandro Ponce-Flores, Colleen B. Jonsson, Daniel N. Streblow, Jennifer E. Golden

TL;DR
Researchers optimized a quinazolinone compound to effectively inhibit Chikungunya virus in human cells, showing promise for future antiviral development.
Contribution
The study reengineered antiviral quinazolinones to achieve potent inhibition of Chikungunya virus with improved cellular activity.
Findings
(R)-1h (BDGR-651) reduced CHIKV titer by 4.1 log at 10 μM in human dermal fibroblasts.
Compound (R)-1h showed excellent solubility and stability in mouse microsomes and plasma.
Confocal microscopy confirmed a dose-dependent protective effect of (R)-1h in infected Vero E6 cells.
Abstract
Chikungunya virus (CHIKV) results in debilitating chronic pain in nearly half of those infected. With no FDA approved small molecule-based therapeutics available, we screened compounds to reveal quinazolinone (S)-1a with a modest 0.3 log reduction of CHIKV titer and no significant toxicity (CC50 > 40 μM). Five scaffold regions were surveyed to improve the titer reduction efficiency. Chemistry was established to preserve the enantiopurity of 2-piperidinyl-containing analogues, affording (R)-1h (BDGR-651) which reduced CHIKV titer in normal human dermal fibroblasts by 4.1 log at 10 μM (EC50 = 0.86 μM). Excellent solubility and mouse microsomal and plasma stabilities were observed, and confocal microscopy of infected Vero E6 cells treated with (R)-1h showed a dose-dependent protective effect. A narrow selectivity index prevented in vivo evaluation, but the study showed that…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
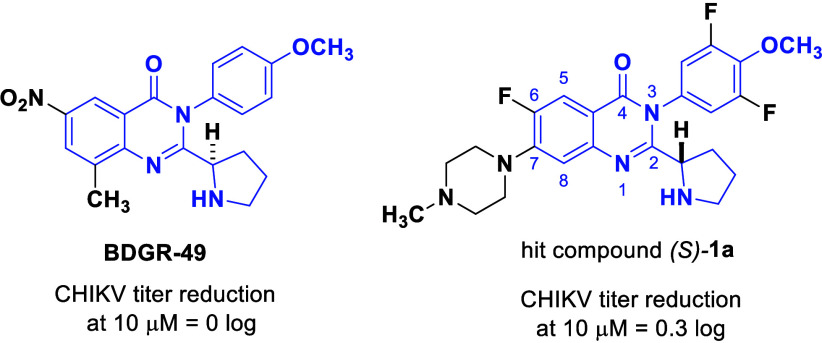 Figure 1
Figure 1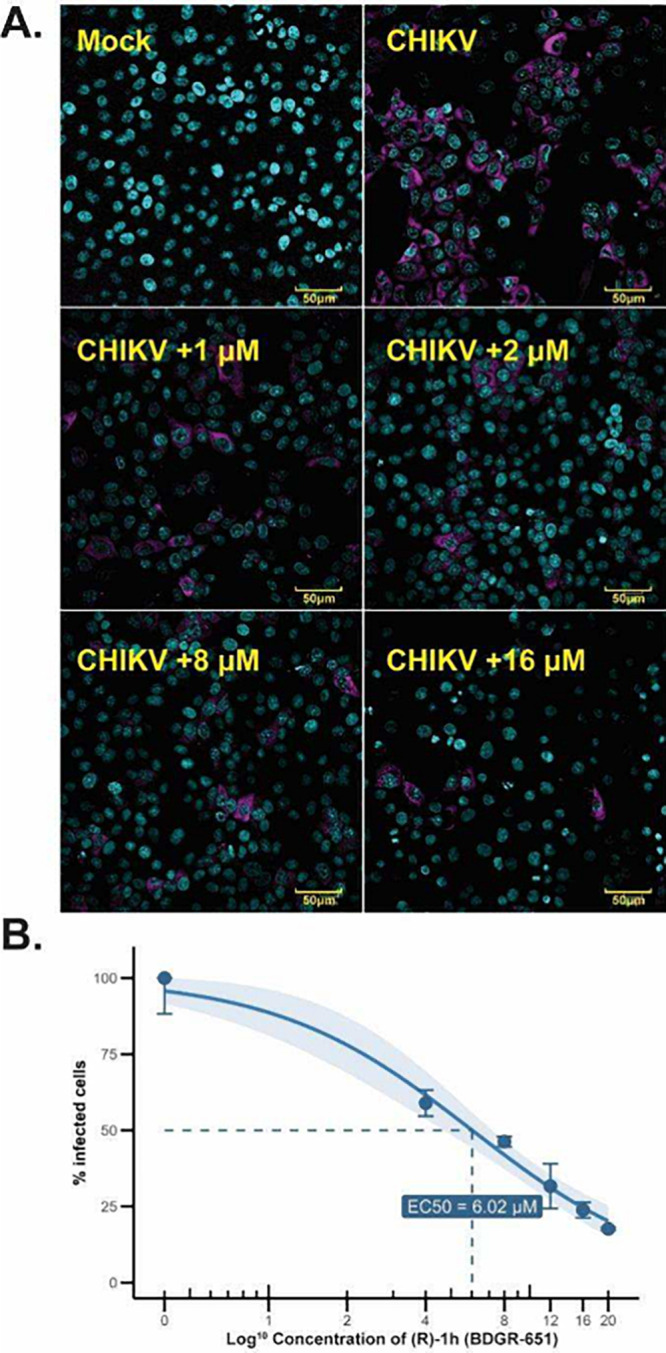 Figure 2
Figure 2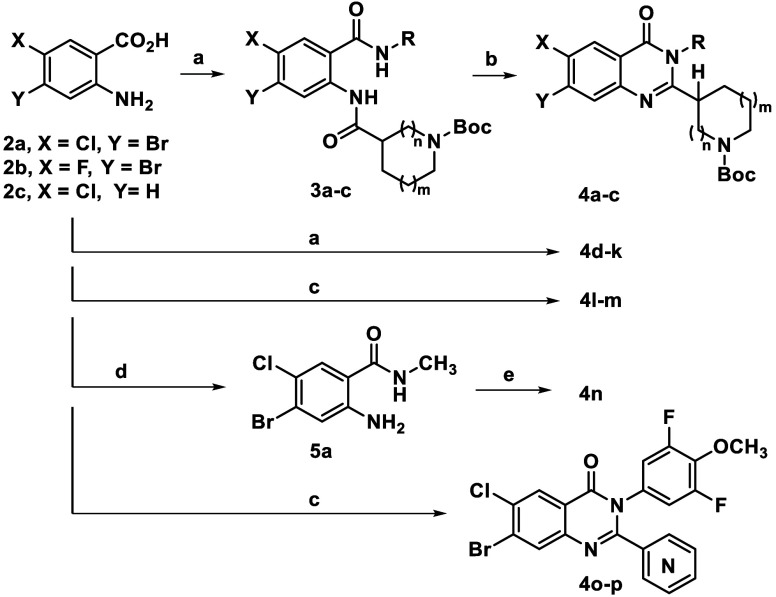 Figure 3
Figure 3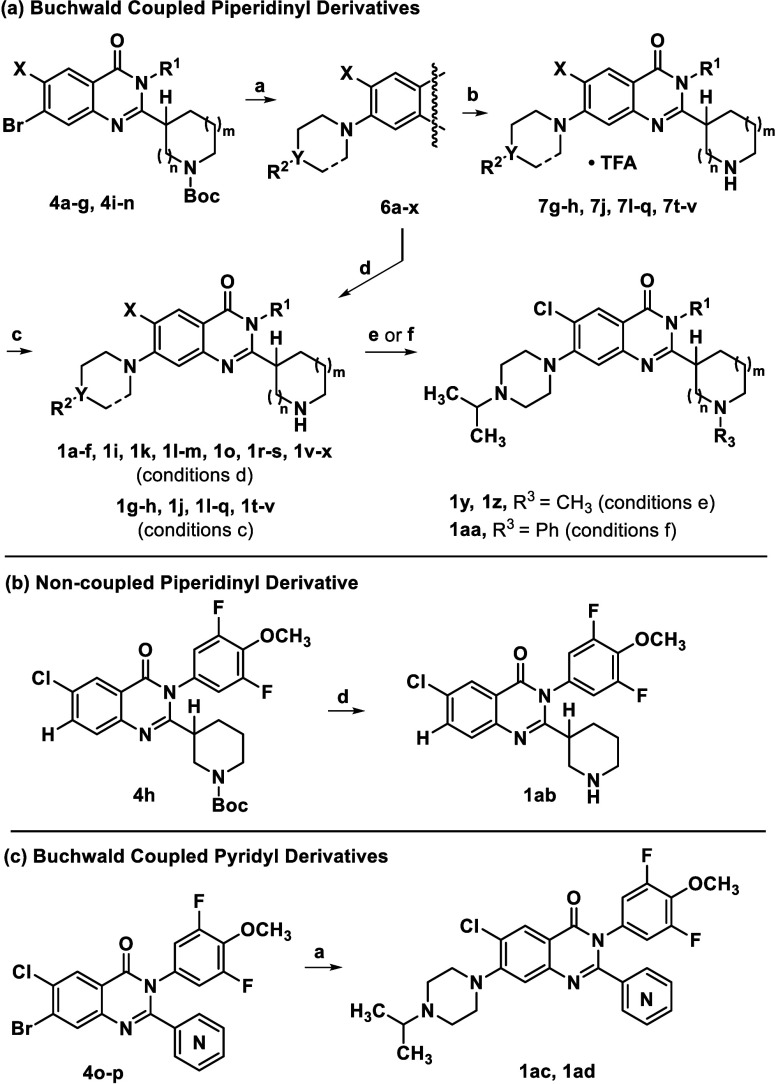 Figure 4
Figure 4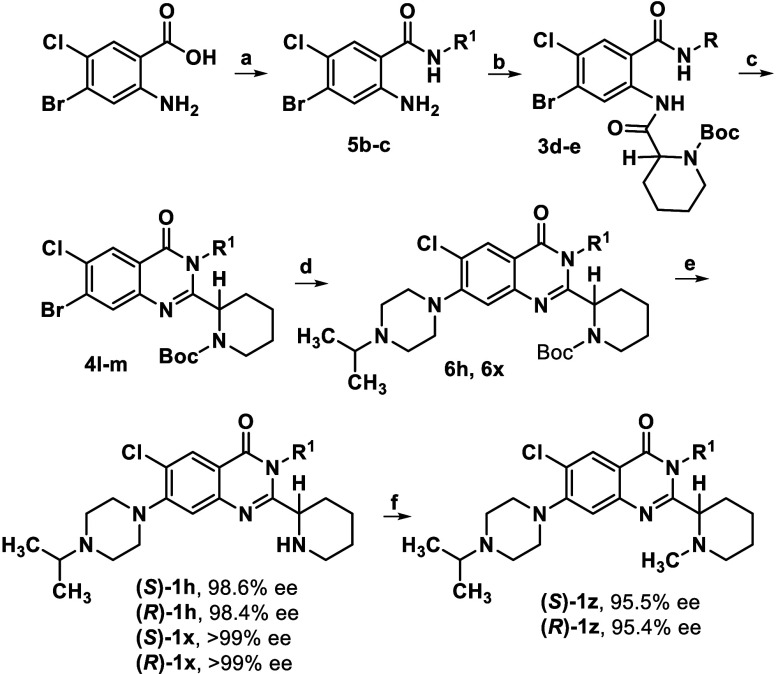 Figure 5
Figure 5 Figure 6
Figure 6 Figure 7
Figure 7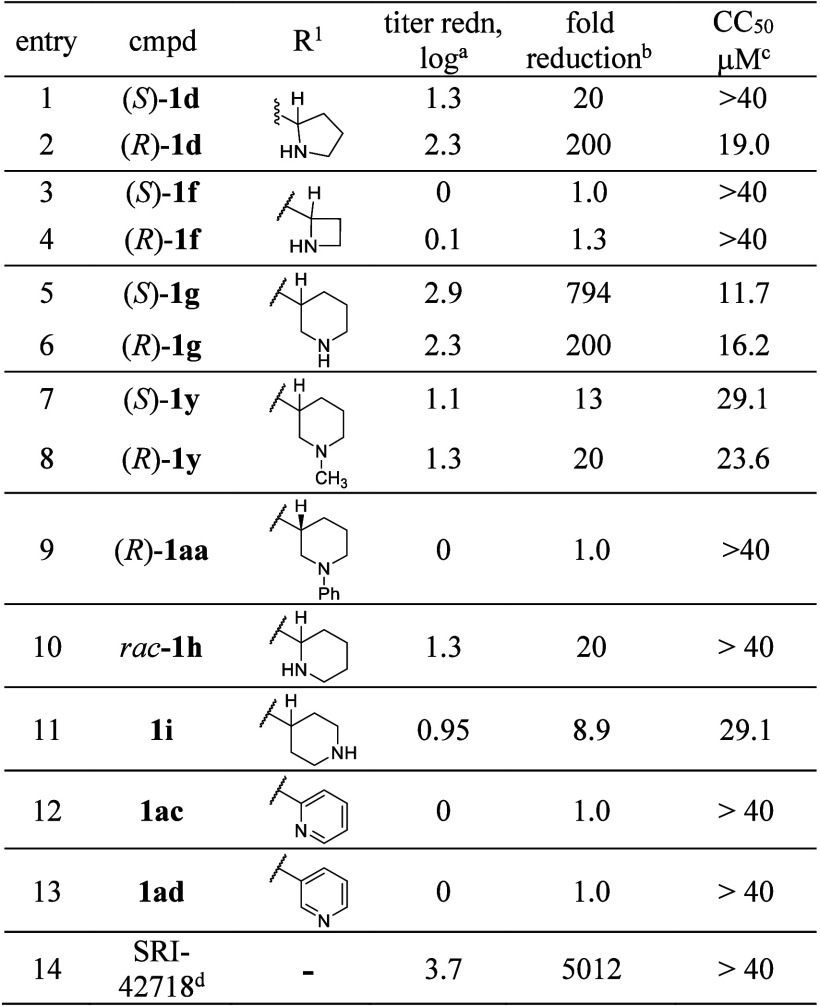 Figure 8
Figure 8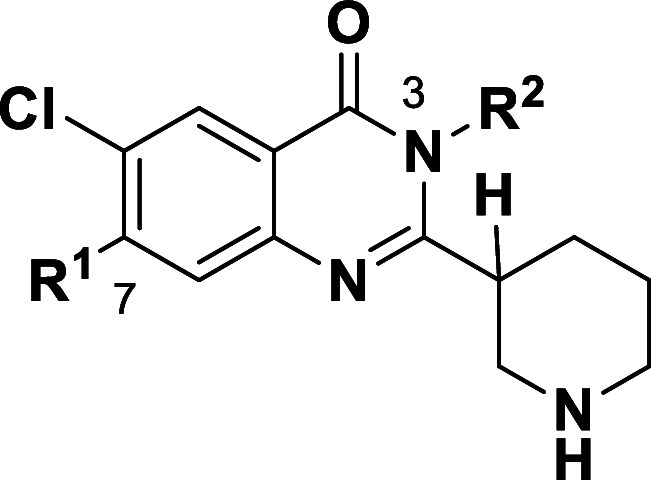 Figure 9
Figure 9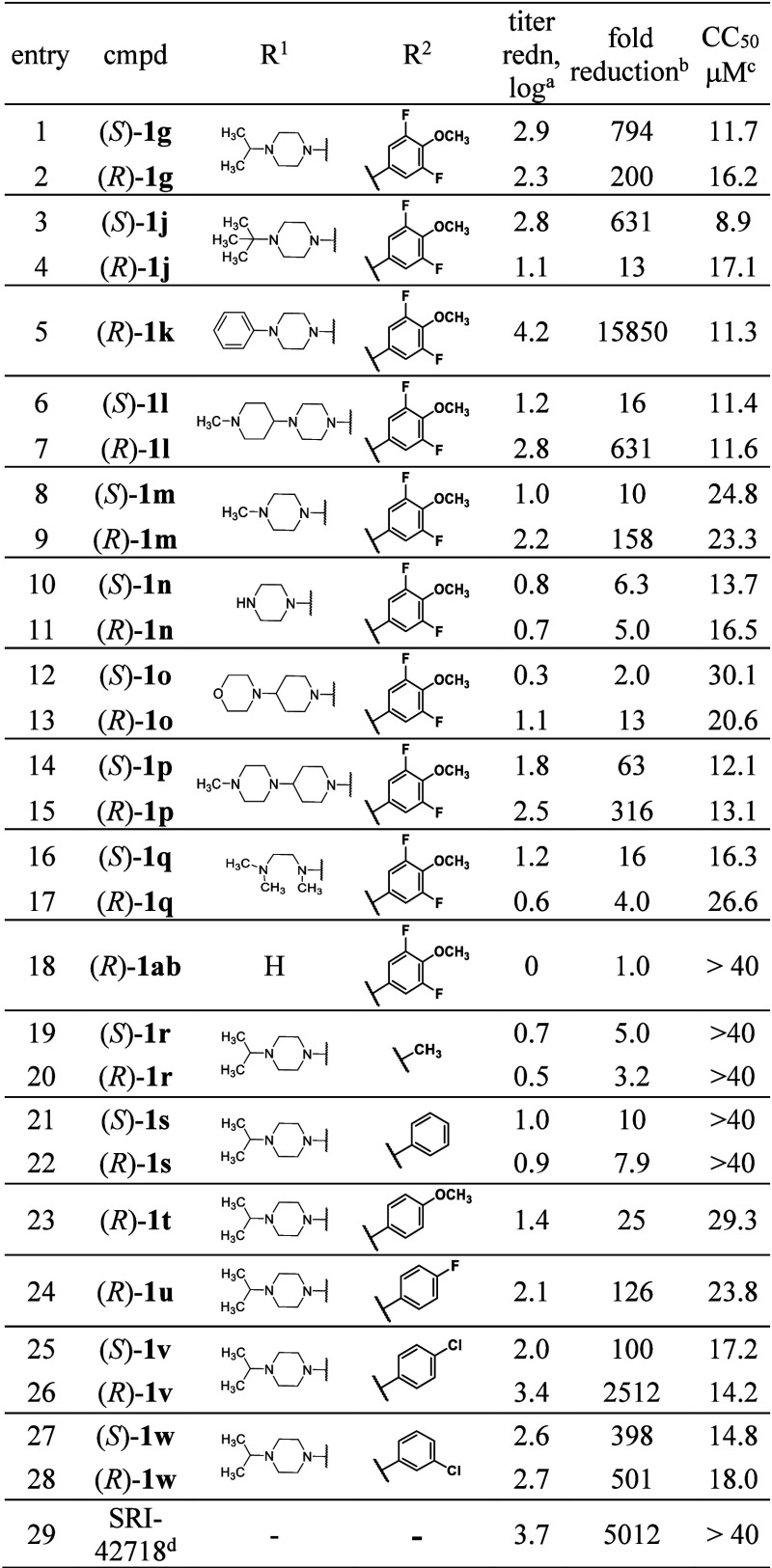 Figure 10
Figure 10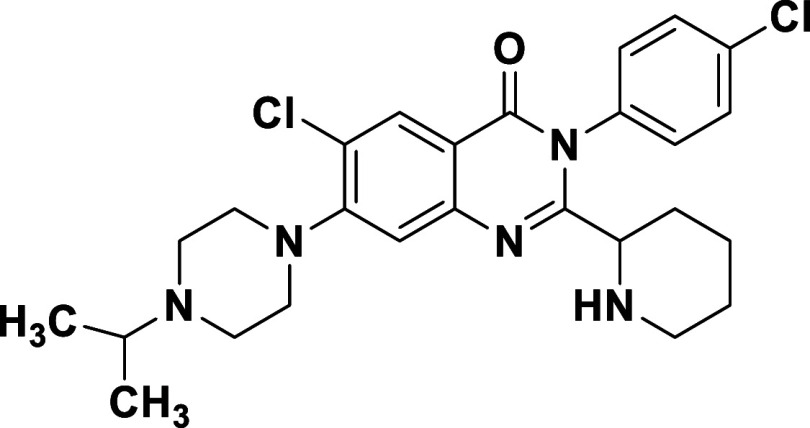 Figure 11
Figure 11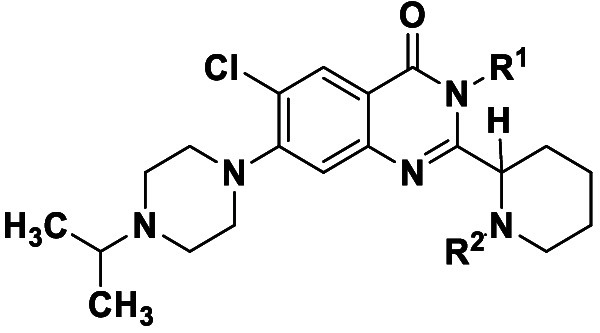 Figure 12
Figure 12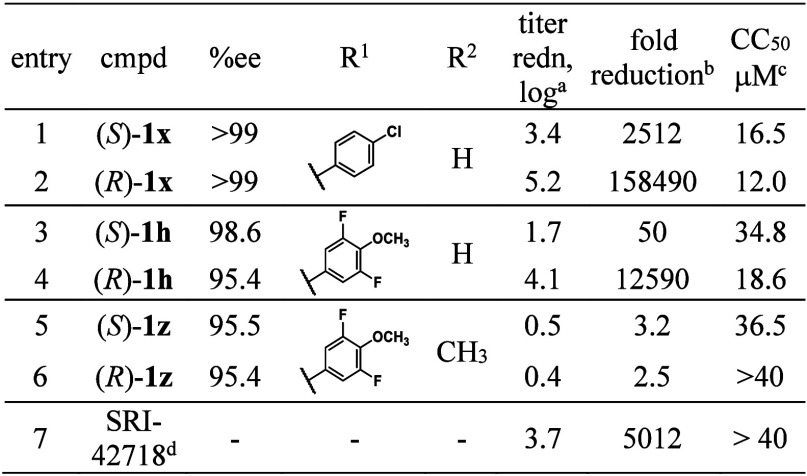 Figure 13
Figure 13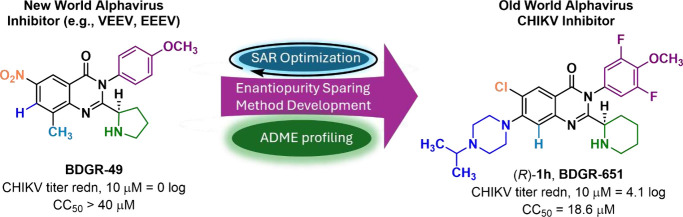 Figure 14
Figure 14 Figure 15
Figure 15- —National Institute of Allergy and Infectious Diseases10.13039/100000060
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMosquito-borne diseases and control · Viral Infections and Vectors · Quinazolinone synthesis and applications
Chikungunya virus (CHIKV) is a mosquito-borne, positive-sense, single-stranded RNA alphavirus. It causes a human illness characterized by severe muscle and joint pain, fatigue, headache, nausea, and rash. ?−? ? ? ? ? Symptoms can resolve within weeks of the initial infection; however, at least 40% of patients suffer from debilitating arthritis pain that persists for months or years. ?,? CHIKV is designated as a priority pathogen due to its rapid emergence, pandemic potential, and the severity of human disease it induces. ?,?,? Currently, two vaccines are available. Ixchiq is a live, attenuated, single-dose vaccine that was FDA approved for those 18 years and older. It is not prescribed for those with weakened immune systems,? and it carries a warning for serious adverse events in older adults (60+ yrs).? Vimkunya, a recombinant single-dose vaccine, was approved in 2025 for use in people of ages 12 and older but also has limitations in pregnant women and the immune-compromised.? For infected individuals, however, only supportive care is available. ?−? ? Global climate changes have enabled mosquito migration and led to the expansion of CHIKV infection beyond endemic areas of Africa and Asia. ?,? More than three-quarters of the world’s population is now at risk,? including frequent travel spots within the Americas, Europe, and territories throughout the Caribbean Sea, as well as the Pacific and Indian oceans. ?,? Thus, the global distribution of CHIKV infection is likely to increase, underscoring the need for safe and effective therapeutics. Given our extensive development of inhibitors ?−? ? ? against encephalitic alphaviruses (e.g., VEEV, EEEV), we screened a selection of our internal compound library to identify potential CHIKV hits. While the quinazolinone BDGR-49 potently inhibited encephalitic alphaviruses, ?,? it showed no appreciable activity against CHIKV at 10 μM (Figure).
Nonetheless, we were intrigued by compounds with a differentiated substitution pattern on the quinazolinone core of BDGR-49 that showed a meager reduction of CHIKV titer at a concentration of 10 μM, exemplified by the hit compound (S)-1a. We decided to explore the distinguishing structural features of compound (S)-1a to establish a potential pharmacophore, improve potency, and investigate preliminary physiochemical and in vitro ADME parameters.
To assemble analogues, a variety of N-Boc protected, quinazolinone intermediates were generated prior to further diversification (Scheme). In all cases, the analogues were built from commercially available anthranilic acids 2a–c. Thereafter, we often employed a one-pot, three step, copper-mediated protocol that was developed in our lab to preserve the stereochemistry of chiral amino acid derivatives used during the quinazolinone core assembly.? For intermediates 4a–c, an additional step using N,O-bis(trimethylsilyl)acetamide was necessary to ring-close the bis-amide precursors 3a–c.? For intermediates 4d–k, the ring closure proceeded directly. For other N3-aryl compounds that did not form under the copper-mediated conditions (e.g., 4l–m) or those lacking a stereocenter to be concerned with, such as 4o–p, harsher conditions using triphenyl phosphite were employed. For compound 4n, which bore an N3-methyl substituent in place of an aryl ring, N-methylamide 5a was formed via coupling of the anthranilic acid,? followed then by ring closure.
Quinazolinone intermediates 4a–g and 4i–n were subjected to Buchwald coupling conditions to afford amine substituted cores 6a–x (Scheme, path a). Removal of the N-Boc group afforded TFA salts 7g-h, 7j, 7l-q, and 7t-v, which were isolated, characterized, and eventually free-based for this project to afford analogues 1g-h, 1j, 1l-q, and 1t-v. More recently generated intermediates were N-deprotected, and after implementing a basic workup, isolated as nonsalts directly to afford 1a-f, 1i, 1k, 1l-m, 1o, 1r-s, and 1v-x. Additional derivatives were obtained using a reductive amination? or coupling procedure to install N-piperidinyl substituents (1y, 1z, 1aa). Piperidinyl analogue 1ab was generated via N-deprotection of the N-Boc intermediate 4h (path b), and pyridyl analogues 1ac and 1ad were obtained via Buchwald coupling of the bromide precursors 4o–p (path c).
The compounds were tested in a cell-based, antiviral titer reduction assay employing CHIKV (strain 181/25mKate) in normal human dermal fibroblast (NHDF) cells. ?,? Each compound was tested in triplicate at 10 μM and compared against a negative DMSO control and an unpublished positive control, SRI-42718, a CHIKV nsP4 polymerase inhibitor.? Plaques were counted using a stereomicroscope at 72 h postinfection. Selected compounds were also evaluated at multiple concentrations to afford EC_50_ values. In parallel, compounds were also assessed for cytotoxicity in NHDF cells at 72 h in a dose response format at a maximum of 40 μM concentration.
Early structural modifications were made to assess stereochemical preferences and the effect of changing the N-substituent on the piperazine moiety. Broad structural exploration of the C6-fluorine atom was challenging due to limited commercial availability or synthetic routes leading to anthranilic acids with both a C6-substituent and neighboring coupling-friendly C7 functionality. As such, the C6 position featured either a fluorine or chlorine atom in all reported analogues. No significant preference for one enantiomer over another was observed in the log titer reduction for either the R- or S-isomer of hit 1a (Table, entries 1–2); however, improved titer reduction was associated with the R-isomer of several analogues in the overall study, depending on the substituents distributed across the scaffold.
A 20-fold improvement in viral titer reduction compared to the control was observed by exchanging the piperazinyl N-methyl group for the more sterically demanding N-isopropyl group (Table, entry 4) while also revealing a preference for the (R)-isomer. Replacement of the C6 fluorine atom with a chlorine atom in the hit structure showed a stronger preference for the (S)-enantiomer (entry 5) and a more pronounced reduction in viral plaques compared to that in the fluorine-containing hit. As such, the effect of integrating the piperazinyl N-isopropyl substituent in tandem with a C6 chlorine atom was surveyed, resulting in the discovery of (R)-1d, which exhibited a 200-fold reduction of CHIKV titer but with some cytotoxicity noted (entry 8). The absence of a piperazinyl alkyl substituent (R^2^ = H) showed similar outcomes for selectivity and enantiomeric preferences (entries 9–10).
While enantiomeric preferences were still unclear, the early results suggested that the piperazinyl N-isopropyl substituent, in combination with a C6 chlorine atom, was preferred over the fluorine atom in terms of virus titer reduction. Synthetically, C6-fluorine containing intermediates would often stall at the bis-amide stage and failed to undergo efficient ring closure, thereby preventing a robust synthesis of C6-fluorine containing analogues. With this in mind, we forged ahead with the SAR studies preserving the C6 chlorine atom and N-isopropyl group while further investigating the role of the C2 pyrrolidine ring and its associated stereochemistry (Table).
The azetidine analogues (Table, entries 3–4) suffered from significant erosion of enantiopurity during synthesis (41–64% ee), but nonetheless, neither offered antiviral advantages over the pyrrolidines, so a variety of six-membered ring azacycles were evaluated. While the 3-piperidine analogue (S)-1g offered a ∼800-fold reduction in CHIKV titer, the increased cytotoxicity associated with this compound obscured a clear understanding of its significance. Alternatively, (R)-1g afforded a profile like that of pyrrolidine (R)-1d. Piperidinyl N-methyl or N-phenyl variants were not advantageous over other modifications (entries 7–9). The 2-piperidine analogues could not be initially synthesized effectively without epimerizing the stereocenter (<10% ee in most examples). To avoid generating a scalemic mixture, racemic 1h was intentionally generated, which showed a 20-fold reduction in viral titer but notably without observed cytotoxicity at 40 μM (entry 10). The 4-piperidinyl 1i and the pyridyl derivatives, 1ac-ad, showed inferior antiviral effects.
We pursued the piperidinyl-substituted analogues given the possibility of improved antiviral activity and the idiosyncratic cytotoxicity observed with various substitution patterns (Table). As discussed later, we also developed a route to 2-piperidyl analogues without sacrificing the enantiopurity of intermediates with the intent of integrating learned SAR optimizations.
We selected the 3-piperidinyl analogue (R)-1g as a template to scout alterations of the C7 N-isopropyl piperazine (Table, entries 3–18). The N-t-butyl piperazine (S)-1j and N-phenyl piperazine (R)-1k showed a nearly 3- and over 4-log reduction of viral plaques, respectively, but with confounding cytotoxicity overlapping in the same concentration window, which prevented a clear interpretation of antiviral activity. Further elongation of the N-substituent to include an N-piperidine led to a similar cytotoxicity outcome. However, the smaller N-methyl piperazine (R)-1m showed a better selectivity profile. Replacement of the alkyl substituent with a hydrogen atom (entries 10–11) led to inferior antiviral activity while also showing cytotoxicity. Elongated piperidine derivatives (entries 12–15) and acyclic analogues (entries 16–17) also did not improve the compound profiles. Replacement of the N-isopropyl moiety with a hydrogen atom (entry 18) abolished all antiviral activity.
While preserving the C7 N-isopropyl piperazine, we also varied the N3-substituent (Table, entries 19–28). Switching from the difluoromethoxyphenyl moiety to a methyl group or a simplified phenyl ring attenuated antiviral activity (entries 19–22). Halogenated phenyl rings fared better, resulting in 2- to nearly 3.5 log reductions in viral titer but still with cytotoxic effects. These structural permutations were conducted in parallel to explorations of a modified synthesis of 2-piperidinyl quinazolinone derivatives, which were racemized in our earlier protocols (see Table, entry 10). We were intrigued by this structural feature as it avoided cytotoxicity and was hypothesized to hold promise when paired with other structural features that reduced viral plaques. In fact, prototype rac-1x (bearing the C7 N-isopropyl piperazine and N3 4-chlorophenyl substituent) was determined to reduce viral titer by nearly 4 log while demonstrating favorable physiochemical and preliminary ADME characteristics, including excellent plasma and microsomal stability in mice (Table).
We employed triphenyl phosphite to assemble 2-piperidinyl containing quinazolinones, but the harsh conditions led to significant epimerization, so we opted to prepare and assess racemates of these analogues initially. While we had previously developed an excellent protocol to address epimerization for most quinazolinones, we noted that the one-pot, copper-mediated method (Scheme, step a) failed to afford the desired product when applied to these 2-piperidinyl analogues. Stepwise analysis of the copper-mediated protocol revealed that the intermediary benzoxazinone S3 did not form, likely due to the presence of the bulky N-Boc-2-piperidinyl substituent (see SI, Scheme S3).
Alternatively, we decided to build the bis-amide intermediate in a stepwise fashion and evaluate condensation methods to form the quinazolinone ring while preserving the stereochemistry component appended to it (Scheme). After extensive screening of various strategies and conditions (SI, Tables S1–S2), we determined that HATU-facilitated formation of benzamides 5b–c was preferential before integrating the more sterically demanding N-Boc-2-piperidinyl substituent (Scheme). The second amidation to form 3d–e was optimally executed using POCl_3_ ? due to the comparatively short reaction time over other options, ease of purification, and retention of starting material and intermediate enantiopurity. Subsequent quinazolinone ring closure was successfully completed to afford 4l–m using N,O-bis(trimethylsilyl)acetamide, thereby providing the best overall results in terms of yield and enantiopurity preservation (Scheme).
Buchwald coupling, followed by N-Boc deprotection, afforded each individual enantiomer of compounds 1h and 1x. The N-methylation of the piperidinyl ring nitrogen atom via reductive amination gave the enantiomers of 1z. This approach, applied to the synthesis of selected analogues that incorporated key functionality at N3 and C6–7, afforded each enantiomer with >95% ee (Scheme). Assessment of the individual isomers of 1h, 1x, and 1z (Table) revealed (R)-1h, known internally as BDGR-651, with the best combination of viral plaque reduction (4.1 log) and cytotoxicity (CC_50_ ∼ 19 μM). The titer reduction assay, performed in dose response format, revealed an EC_50_ = 0.86 μM in NHDF cells (Figure S1).
The effect of (R)-1h on CHIKV infection was observed microscopically using high-content confocal imaging of the viral capsid within infected Vero E6 cells (FigureA). Uninfected, untreated cells stained brightly with nuclear staining (mock control) and were abundant. Cells infected with CHIKV at an MOI of 1 without (R)-1h showed a reduced number of stained cells. The addition of (R)-1h in increasing concentrations to infected cells at 1, 2, 8, 16, and 20 μM resulted in a dose-dependent protective effect in which the highest concentration of (R)-1h reduced CHIKV induced cell death by the greatest degree. Quantitative assessment of the capsid protein in 20 images per well in triplicate for all concentrations revealed an EC_50_ = 6.0 μM (FigureB). At the highest compound concentrations in Vero E6 cells, cytotoxic effects were observed for (R)-1h, resulting in a decrease in live cell count. The CC_50_ value in this cell line was determined to be >20 μM.
While the selectivity index of (R)-1h limited its advancement into pharmacokinetic models, quinazolinone (R)-1h (BDGR-651) was profiled for initial physiochemical and ADME properties to characterize and benchmark these attributes as a function of structural features prior to optimizing the series further. Compared to the early prototype, rac-1x (Table), a 5-fold boost in aqueous solubility and a significant free fraction of unbound compound in mouse plasma was observed for (R)-1h (Table). Gratifyingly, excellent mouse plasma and microsomal stability was preserved. On these grounds alone, (R)-1h displayed a reasonable profile which suggests that further tuning of potency and cytotoxicity may bode well for the series in additional rounds of optimization. This will be the subject of future work involving this CHIKV-based quinazolinone series.
Despite the extensive quinazolinone compound library built internally to combat New World encephalitic alphaviruses such as VEEV and EEEV, no significant activity was observed for selected compounds that were tested against the Old World arthritogenic alphavirus CHIKV. Nonetheless, assessment of a broader sampling of quinazolinones from our collection that were negative controls against the encephalitic alphaviruses revealed modest anti-CHIKV activity in a titer reduction assay. As a result, an optimization effort involving five regions of the quinazolinone core improved the titer reduction capability from a meager 0.3 log of hit compound (S)-1a to an analogue, (R)-1h, that reduced CHIKV plaques by 10,000-fold compared to the DMSO control while also exhibiting favorable physiochemical and tier one ADME characteristics.
Key lessons from the SAR effort included a preference for a C5 substituent combined with a fluorine or chlorine atom at C6. The presence of a substituted aryl ring on the quinazolinone core N3 position was preferred over a simple phenyl ring or methyl group, and 2-piperidines at C2 were favored over four-, five-, and other isomeric six-membered ring amines. A strong stereochemical preference regarding the C2 moiety was not apparent, although combinatorial structural changes across the scaffold appear to influence enantiomeric preferences. Compound (R)-1h featured the presence of a quinazolinone C2 2-piperidinyl moiety, the installation of which required the development of an alternative synthetic strategy to successfully form the quinazolinone while also preventing racemization of the stereochemistry of the partnered amino acid derivative.
Some degree of cytotoxicity was observed for most analogues in this series due to structural differences – contrary to that observed for the VEEV and EEEV inhibitors, and generally, the magnitude of the cytotoxicity may correlate with antiviral effects. However, some analogues showed a broader selectivity index that suggested that a more extensive SAR effort, especially focused on the N3 aryl substituent and C7 and C8 substitution, may offer opportunities for improvement. These future studies will be important to pursue resistant mutations to reveal potential involvement of viral proteins, studies that were hampered in the current work by the low selectivity index with (R)-1h, as well as assessing in vivo pharmacokinetic profiles and efficacy, which are premature at this time. Compared to quinazolinones like BDGR-49 that potently inhibit VEE and EEE alphaviruses, CHIKV quinazolinones contain pivotal structural differences (e.g., C6 nitro group to halide change) that are inconsistent with quinazolinone SAR that is known to be required for encephalitic alphavirus inhibition. As such, we suspect that the mechanism of action of the CHIKV quinazolinones may be different. This will be assessed and reported elsewhere. Nonetheless, this work highlights the capacity of the quinazolinone core to be structurally tuned for activity against phylogenetically related yet unique viruses such as VEEV and CHIKV which make targeting them effectively with a single compound particularly challenging. The development of chemical scaffolds that can be optimized for re-emerging viruses such as CHIKV is critical, as the effects of global warming remain significant and variably unmitigated. Further, new therapies will be needed to address the potential increased number of infections and vulnerable populations.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abdelnabi R.Jacobs S.Delang L.Neyts J.Antiviral drug discovery against arthritogenic alphaviruses: Tools and molecular targets Biochem. Pharmacol.202017411377710.1016/j.bcp.2019.11377731874146 · doi ↗ · pubmed ↗
- 2Abdelnabi R.Delang L.Antiviral Strategies against Arthritogenic Alphaviruses Microorganisms 202089136510.3390/microorganisms 809136532906603 PMC 7563460 · doi ↗ · pubmed ↗
- 3Suhrbier A.Jaffar-Bandjee M.-C.Gasque P.Arthritogenic alphavirusesan overview Nat. Rev. Rheumatol.20128742042910.1038/nrrheum.2012.6422565316 · doi ↗ · pubmed ↗
- 4Oliveira Silva Martins D.De Andrade Santos I.Moraes De Oliveira D.Riquena Grosche V.Carolina Gomes Jardim A.Antivirals Against Chikungunya Virus: Is the Solution in Nature?Viruses 202012327210.3390/v 1203027232121393 PMC 7150839 · doi ↗ · pubmed ↗
- 5Heise, M. ; Frolov, I. ; Frolova, E. I. ; Carissimo, G. ; Ng, L. F. P. ; Mc Carthy, M. K. ; Davenport, B. J. J. ; Morrison, T. E. ; De Filippis, V. R. ; Haese, N. ; Powers, J. ; Streblow, D. N. In Chikungunya Virus; Heise, M. , Ed.; Current Topics in Microbiology and Immunology, Vol. 435; Springer International Publishing: Switzerland, 2022.
- 6Bassetto, M. ; Brancale, A. The search for antivirals to treat alphavirus infections. In Annu. Rep. Med. Chem. Vol. 57; Elsevier, 2021; pp 133–151.
- 7De Souza W. M.Ribeiro G. S.De Lima S. T. S.De Jesus R.Moreira F. R. R.Whittaker C.Sallum M. A. M.Carrington C. V. F.Sabino E. C.Kitron U.Chikungunya: a decade of burden in the Americas Lancet Reg. Health Am.20243010067310.1016/j.lana.2023.10067338283942 PMC 10820659 · doi ↗ · pubmed ↗
- 8Amaral J. K.Bingham C. O.Taylor P. C.ViláL. M.Weinblatt M. E.Schoen R. T.Pathogenesis of chronic chikungunya arthritis: Resemblances and links with rheumatoid arthritis Trav. Med. Infect. Dis.20235210253410.1016/j.tmaid.2022.102534 · doi ↗