Novel Mutations in Titin Exon 363 With Different Phenotypes Including a Founder Mutation in Eastern Europe
Veronica Sian, Maria Francesca Di Feo, Sergei Kurbatov, Anna Vihola, Helena Luque, Fedor Konovalov, Stojan Peric, Cathrina Duffy, Cornelia Kornblum, Kristl G. Claeys, Peter Hackman, Bjarne Udd, Marco Savarese

TL;DR
The study identifies new mutations in the TTN gene's exon 363 that cause different types of muscle disease, including a common mutation in Eastern Europe.
Contribution
The study reports two novel pathogenic TTN exon 363 variants causing recessive titinopathies with distinct clinical features.
Findings
A founder mutation in exon 363 is associated with juvenile distal titinopathy in Eastern Europe.
A frameshift deletion in exon 363 causes congenital myopathy with contractures in a Belgian family.
Disease severity depends on the location of the second TTN variant.
Abstract
Titin, the largest human protein, is essential for sarcomere structure and function. The TTN gene, spanning 364 exons, undergoes extensive alternative splicing thus producing multiple isoforms. The M‐band region, encoded by exons 359–364, plays a critical role in sarcomere integrity and mechanical stability. Exon 363 is of interest due to its involvement in titinopathies. Pathogenic truncating variants in this exon have been linked to recessive myopathies, including and mainly young‐onset recessive distal titinopathy. A multicenter study was conducted on six patients from five unrelated families with confirmed recessive titinopathy and truncating variants in exon 363. Clinical evaluations were performed. Genetic testing and segregation analysis confirmed the phase of the variants. A novel truncating variant c.107578C>T, p.(Gln35860Ter) was identified in four unrelated patients of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
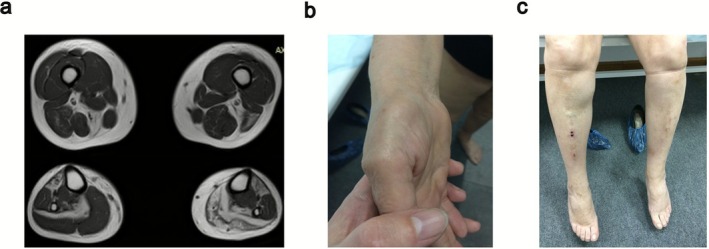 FIGURE 1
FIGURE 1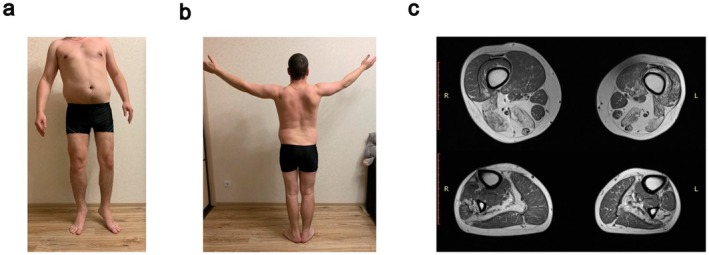 FIGURE 2
FIGURE 2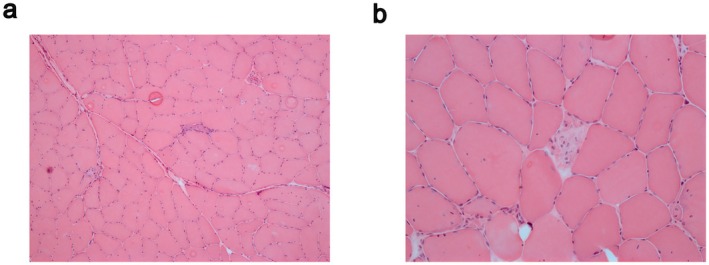 FIGURE 3
FIGURE 3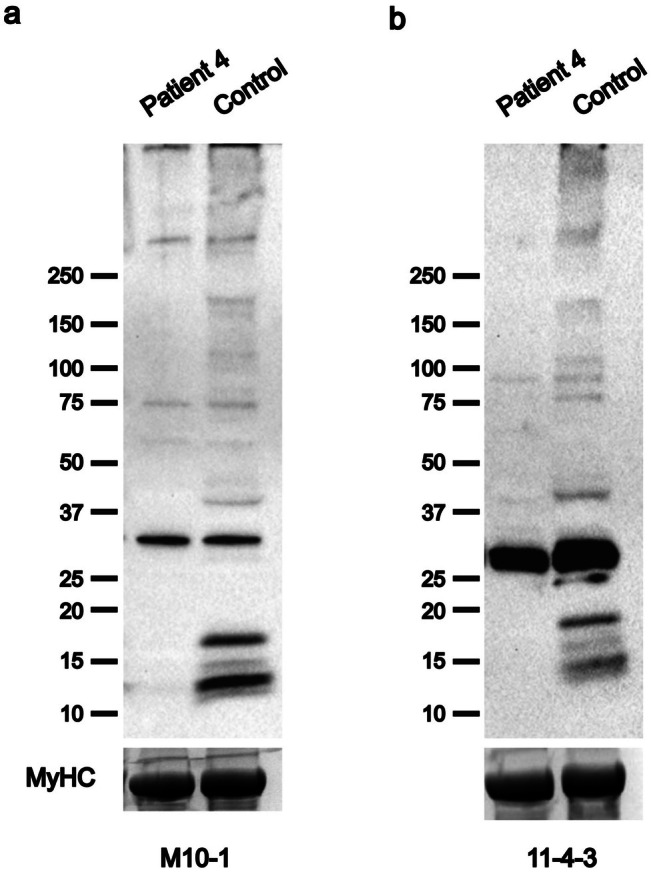 FIGURE 4
FIGURE 4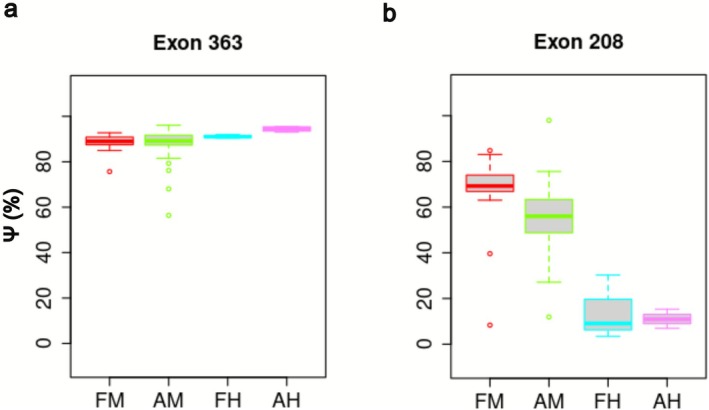 FIGURE 5
FIGURE 5| ID | Sex | Age at onset | Age at last examination | Symptomps at onset | Independent walking | Weakness distribution | Cardiac involvement | Respiratory involvement | Muscle biopsy report | CK | EMG | Muscle MRI | Other | TTN var 1 | Exon var 1 | TTN var 2 | Exon var 2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 23 | 46 | LL muscle wasting and muscle weakness | Yes | LL > UL | No | No | Rimmed vacuoles, fiber type variation, internal nuclei | Normal‐mildly elevated | NA | NA | Asymmetry |
c.107578C>T p.(Gln35860Ter) | 363 |
c.50228_50229del p.(Val16743AlafsTer19) | 267—A‐band |
| 2 | F | 26 | 58 | LL muscle wasting and muscle weakness | Yes (steppage gait) | LL > UL | No | No | NA | Normal‐mildly elevated | Myogenic | Fatty degeneration of the right Adductor magnus and left semitendinosus, biceps femoris. Severe fatty replacement in soleus and anterior compartment muscles with gastrocnemius involvement on the left side | Asymmetry, thenar atrophy since the age of 50 |
c.107578C>T p.(Gln35860Ter) | 363 |
c.66720C>G p.(Tyr22240Ter) | 317—A‐band |
| 3 | M | 20 | 37 | Running difficulties | Yes | LL > UL | No | No | Autophagic vacuoles | Normal‐mildly elevated | Myogenic | Fatty degeneration of hamstrings and Adductor magnus and hypotrophy of left quadriceps on the thigh. Severe fatty replacement in soleus and tibialis anterior muscles | Asymmetry, rigid spin, reduced tendon reflexes |
c.107578C>T p.(Gln35860Ter) | 363 |
c.106571dup p.(Thr35525Aspfs*3) | 361—M‐band |
| 4 | M | 34 | 42 | Walking difficulties | Yes | LL > UL | No | No | Prevalence of type 1 fiber, fiber splitting, increased central nuclei, vacuoles, degenerating, and regenerating fibers, a mild endomysial infiltrate (CD‐68) and mild inflammatory reaction | Mildly elevated | NA | Severe fatty changes in the anterolateral and soleus muscles of the lower legs | Calf pseudohypertrophy |
c.107578C>T p.(Gln35860Ter) | 363 |
c.107840T>A p.Ile35947Asn | 364—M‐band |
| 5_1 | M | Childhood | 51 | Walking difficulties, contractures | No | LL > UL | No | Yes | Dystrophic features | Normal‐mildly elevated | Myogenic | No | NA |
c.107431del p.(Ser35811AlafsTer33) | 363 |
c.39524del p.(Lys13175SerfsTer22) | 208—A‐band |
| 5_2 | M | Childhood | 48 | Walking difficulties, contractures | No | LL > UL | No | Yes | NA | Normal‐mildly elevated | NA | No | NA |
c.107431del p.(Ser35811AlafsTer33) | 363 |
c.39524del p.(Lys13175SerfsTer22) | 208—A‐band |
| Position (hg19) | ExonicFunc | AAChange | Exon |
|---|---|---|---|
| 179392275 | stopgain | c.107578C>T:p.Q35860X | 363 |
| 179424048 | synonymous_SNV | c.86811A>G:p.V28937V | 327 |
| 179498042 | nonsynonymous_SNV | c.42958A>G:p.K14320E | 234 |
| 179583317 | synonymous_SNV | c.24516C>T:p.T8172T | 86 |
| 179586604 | nonsynonymous_SNV | c.22786G>C:p.D7596H | 79 |
| 179638238 | synonymous_SNV | c.7545C>T:p.Y2515Y | 32 |
- —Academy of Finland10.13039/501100002341
- —European Union (Compass‐NMD)10.13039/501100000780
- —Samfundet Folkhälsan i Svenska Finland
- —European Joint Program on Rare Diseases (project IDOLS‐G)
- —Sigrid Juséliuksen Säätiö10.13039/501100006306
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiomyopathy and Myosin Studies · Nuclear Structure and Function · Cellular transport and secretion
Introduction
1
Titin is the largest known protein in the human body, playing fundamental structural and functional roles in the sarcomere [1]. Encoded by the TTN gene, titin extends from the Z‐disk to the M‐band within the sarcomere [2, 3], acting as a molecular spring that contributes to muscle extensibility and mechanical stability [4]. The TTN gene consists of 363 coding exons and an additional first noncoding exon [5], and undergoes extensive alternative splicing [4]. The theoretical isoform incorporating all predicted TTN exons corresponds to the inferred full TTN “metatranscript” (NM_001267550.2) [4].
The C‐terminal, M‐band region of titin is encoded by the last 6 exons (359–364) [6]. M‐band titin consists of a serine/threonine kinase (TK) in the large M1 domain, followed by a series of alternating immunoglobulin domains (M1–M10) and unique insertion sequences (is1–is7) [6]. The second last TTN exon 363, which encodes the is7 domain in the M‐band, undergoes differential splicing, resulting in the is7− and is7+ isoforms [7, 8].
The M‐band plays a crucial role in the structural and mechanical stability of myofibrils, particularly regulating thick filament assembly [9]. It serves as a hub for protein–protein interactions, linking titin to myomesin, obscurin, and other key sarcomeric components [10].
Pathogenic variants in the M‐band segment of titin have been implicated in several muscle disorders with or without cardiomyopathies [4, 11]. Several truncating variants in exon 363 have been described so far, including a founder mutation in the Serbian population causing a juvenile onset recessive distal titinopathy [12, 13, 14, 15, 16]. Truncating variants in exon 363 result in a shorter titin and the lack of the C‐terminal portion most likely alters the mechanical and biochemical properties of the protein in muscle tissues [11, 17].
In the present study, we identified two novel nonsense variants in TTN exon 363. In a well‐defined cohort of patients with Eastern European ancestry, we identified a novel founder mutation, a nonsense variant c.107578C>T, p.(Gln35860Ter), causing young‐onset recessive distal titinopathy in trans with pathogenic variants in canonical exons. In a Belgian family, an early‐onset progressive Emery‐Dreifuss‐like titinopathy is caused by a previously unreported deletion c.107430delA, p.(Ser35811AlafsTer32) in exon 363 in compound heterozygosity with a single nucleotide deletion c.39524del, p.(Lys13175SerfsTer22) in exon 208.
Materials and Methods
2
Patients
2.1
All the patients or their legal guardians as well as the patient's relatives provided written informed consent to the referring clinician. The study, approved by the ethics committee of the Helsinki University Hospital (HUS/16896/2022), was performed in accordance with the Declaration of Helsinki.
In this multicenter study, we recruited six patients from five unrelated families with a confirmed recessive titinopathy and a truncating variant in exon 363. Electrophysiological examination results (nerve conduction studies and needle electromyogram (EMG)), creatine kinase (CK) measurements, histological and histochemical muscle examination results, and cardiac function test results were available for most patients (Table 1). Genetic testing was performed by a comprehensive gene panel and/or exome sequencing, and the phase of the variants was confirmed by segregation analysis. All variants are reported according to Human Genome Variation Society recommendations using the inferred complete TTN metatranscript as reference (NM_001267550.2). Exons are numbered 1–364 according to the LRG numbering as adopted by the Leiden Open Variation Database (LOVD, https://databases.lovd.nl/shared/genes/TTN).
Exon usage (expressed as PSI, percentage spliced in) has been retrieved by an online resource (gacatag.shinyapps.io/TTN_PSIVIS/) and calculated as reported previously [18].
Western Blotting
2.2
Frozen muscle biopsies were used to prepare Western blot samples as described before [11, 19]. Samples were run on SDS–PAGE gels (12%) using Bio‐Rad Mini‐Protean equipment (Bio‐Rad, Hercules, CA), and transferred onto polyvinylidene difluoride (PVDF) membranes. Two previously described in‐house–generated antibodies (rabbit polyclonal antibody M10‐1 [11] and mouse monoclonal antibody 11‐4‐3 [19]) were used to detect the titin via the M10 domain, followed by horseradish peroxidase–conjugated secondary antibodies (Dako) and enhanced chemiluminescent detection using the Pierce SuperSignal West Femto substrate (Thermo Fisher).
Results
3
Juvenile‐Young Adult Onset Recessive Distal Titinopathy
3.1
Targeted next‐generation sequencing of myopathy‐related genes identified a novel truncating variant c.107578C>T, p.(Gln35860Ter) in four unrelated patients with a juvenile–young adult‐onset distal myopathy. The four patients, one is from Serbia, two patients are from Russia and the fourth one from Germany, share the same causative variant. All the patients carry a second pathogenic variant in a canonical TTN exon, respectively c.50228_50229del p.(Val16743AlafsTer19) in exon 267 (A‐band), c.66720C>G p.(Tyr22240Ter) in exon 317 (A‐band), c.106571dup p.(Thr35525AspfsTer3) in exon 361 (M‐band), and previously reported missense c.107840T>A p.(Ile35947Asn) in exon 364 (M‐band). The same TTN haplotype was observed for patients 1, 2 and 4, for whom raw genetic data was available (Table 2).
All the patients have been on regular follow‐up for many years, with a minimum of 8 years in Patient 4 and a maximum of 32 years in Patient 2. In all cases, a slowly progressive course was reported. The average age at the last evaluation was 46 years (range 37–58), and all patients were still ambulant.
Patient 1 showed the first symptoms at the age of 23 years. He developed right limb's muscle atrophy, with muscle weakness more pronounced in the lower limbs than in the upper limbs. Prominent asymmetric muscle weakness and atrophy were observed in the right part of the body. Muscle biopsy (no longer available) evidenced myopathic changes with rimmed vacuoles, increased fiber type variation, and internal nuclei. At last examination, at the age of 46, the patient was still able to walk independently, and cardiac and respiratory involvement were ruled out.
Patient 2 showed muscle wasting in the lower leg with consequent muscle weakness at the age of 26 years. She used to be an active person and walk long distances until her twenties. Fatigue and weakness in the lower legs (more on one side), worsened over time and steppage gait has been present from the age of 28. Mild weakness of neck flexors was also noticed, and CK levels were mildly elevated (4×ULN). EMG showed myopathic anomalies, with no spontaneous activity, and MRI at the age of 58 displayed asymmetric muscle atrophy, with diffuse fatty replacement in the left leg, and mostly in the anterior part of the right leg (Figure 1a). Thenar hypotrophy and mild hypothenar hypotrophy (left > right) were also evident (Figure 1b), together with lower leg involvement with both peroneal and tibial weakness (Figure 1c). No respiratory or cardiac involvement was reported at the age of 58.
Main clinical findings in Patient 2. (a) Muscle MRI of the lower limbs showing less severe changes in the thigh with fatty degeneration on the right in Adductor magnus and semitendinosus, biceps femoris on the left side, and the typical early changes observed in juvenile onset recessive distal titinopathy with severe fatty replacement in soleus and anterior compartment muscles (in this case, also asymmetric gastrocnemius involvement on the left side). (b) Thenar and mild hypothenar hypotrophy. (c) Distal lower leg weakness.
Patient 3 had an early onset at the age of 20 years, reporting slight weakness in his left leg while running. At the last examination, at the age of 37, he showed clear asymmetrical atrophy of the lower limbs, with more pronounced involvement of the left leg (Figure 2a), and mild asymmetrical scapular winging (Figure 2b). Rigid spine and reduced tendon reflexes were also observed. Cardiac ultrasound and forced vital capacity were normal. CK values showed fluctuations over the years (between 4×ULN and 6×ULN), without a clear correlation with the clinical symptoms. At the age of 30, muscle MRI showed involvement of the tibial anterior, soleus and peroneal muscles in the distal lower limbs, and hamstrings, vastus intermedius and vastus medialis in the thigh (Figure 2c).
Main clinical findings in Patient 3. (a) Asymmetric lower limb atrophy, left > right. (b) Mild asymmetric scapular winging. (c) Muscle MRI of the lower limbs showing fatty degeneration of hamstrings and adductor magnus and hypotrophy of the left quadriceps on the thigh. And the typical changes in juvenile onset recessive distal titinopathy with severe fatty replacement in soleus and tibialis anterior muscles (in this case, also asymmetric peroneus involvement).
Patient 4 reported, at age 34, difficulties in walking with muscle weakness predominant in the lower limbs. Absence of cardiac and respiratory involvement was observed in the patient at age 42 years. Muscle MRI revealed severe fatty changes in the anterolateral and soleus muscles of the lower legs. The hamstring muscles were the main severely affected muscles of the thighs, and gluteus medius and minimus were the most affected in the pelvic. Muscle biopsy performed at the age of 37 from the left gastrocnemius showed predominance of type 1 fibers, fiber splitting, increased central nuclei, vacuoles, some degenerating and regenerating fibers, a mild endomysial infiltrate (CD‐68) and mild inflammatory reaction without clear signs of myositis. Hematoxylin and Eosin (H&E) staining (Figure 3a,b) evidenced fiber size variation, fibers with increased central nuclei, few atrophic fibers as well as degenerating and regenerating fibers, vacuoles and fiber splitting. His father, carrier of the Belgian Tibial Muscular Dystrophy (TMD)‐causing variant c.107840T>A p.(Ile35947Asn) in exon 364 [20], showed bilateral foot drop at the age of 75, with unsteady and steppage gait. Western blotting analyses showed a marked reduction of small C‐terminal titin protein fragments, indicating a decrease in their expression (Figure 4a,b).
Histologic examination of muscle biopsy. Hematoxylin and eosin (H&E) staining of muscle biopsy from left gastrocnemius of Patient 4. Magnification: (a) 10×, (b) 20×.
Western blot for C‐Terminal Titin Fragments. Western blotting using two different antibodies, (a) M10‐1 and (b) 11‐4‐3, against the titin C‐terminal M10 domain. Patient 4 with a single identified protein truncating variant shows a severe reduction of titin C‐terminal fractions. The myosin heavy chain (MyHC) serves as the loading control.
Early Onset Progressive Titinopathy With Contractures
3.2
Patients 5a and 5b, two affected brothers with an early onset, were found compound heterozygous for a frameshift variant c.107431del p.(Ser35811AlafsTer33) in exon 363 and a second single nucleotide deletion c.39524del p.(Lys13175SerfsTer22) in exon 208 (A‐band).
They both presented with contractures, proximal weakness, and atrophy as with Emery‐Dreifuss‐like phenotype without cardiomyopathy. One brother had a more severe clinical presentation leading to wheelchair confinement since adolescence (currently 51 years), while the other was wheelchair bound since age 25 years (currently 48 years). Cardiac examinations were normal. Lung function tests at the current age of 48 and 51 years showed restrictive lung disease, with FVC 68% (significantly reduced below 80% since age 46 years) and 31% (unknown since what age significantly reduced; no NIV), respectively.
Discussion
4
Among the many exons that encode Titin, exon 363 has drawn particular interest due to its localization within the M‐band and its role in modulating Titin's structural properties [11]. Exon 363 encodes a segment of the Titin protein that is crucial for sarcomere integrity and contributes to the functional specialization of different muscle types [21, 22]. In addition, the inclusion or exclusion of exon 363 through alternative splicing results in distinct Titin isoforms may influence muscle mechanical resilience (Figure 5a) [18, 23].
Exon usage of TTN. PSI box plot representation of (a) exon 363 and (b) exon 208 of TTN in prenatal (red) and postnatal (green) skeletal muscles. AH, postnatal heart; AM, postnatal muscle; FH, fetal heart; FM, fetal muscle.
Despite being an alternatively spliced exon, pathogenic variants affecting exon 363 have been identified in many titinopathy patients [13]. A common founder mutation c.107635C>T p.(Gln35879Ter) has been identified in patients of Serbian origin showing a recessive distal myopathy with a juvenile/early‐adult onset [15]. Similarly, we here report a novel founder mutation c.107578C>T, p.(Gln35860Ter), once again in the Eastern European population, causing a similar distal titinopathy. The variant has a frequency of 0.002338% in the gnomAD European non‐Finnish population and it has been observed in a single allele in the patient subgroup of a Russian database (http://ruseq.ru, total number of alleles = 9550), a large‐scale reference database of genetic variants specific to the Russian population [24].
In the first three patients, the combination of the novel founder mutation and truncating variants in canonical exons results in a similar phenotype due to the complete lack of a titin protein expressing the entire M9 as demonstrated by the strong reduction/absence of C‐terminal fragments in the Western blot in Patient 4 (Figure 4a,b). In addition to the exon 363 variant, Patient 4 has a previously reported pathogenic missense variant in exon 364 p.(Ile35947Asn) [20]. The missense variant in the last exon causes a reduction in C‐terminal fragments due to aberrant proteolytic processing of the titin C‐terminus [16]. Interestingly, Patients 5a and 5b show a more severe phenotype with an early onset progressive titinopathy with contractures, and wheelchair confinement already in adolescence/early adulthood. Although we still lack a clear genotype–phenotype correlation, other congenital/infantile cases with a truncating variant in the last two exons have been reported [19, 25]. The specific location of the second truncating variant and the PSI of the mutated exon explain the phenotype. In patients 1–4, the second variants are located in different exons that have a PSI comparable to that of exon 363 (i.e., over 80% in fetal and postnatal skeletal muscles) (Table S1). On the contrary, in family 5, the second variant is located in exon 208 showing a PSI of 67% in fetal muscles and 56% in postnatal muscles (Figure 5b).
In conclusion, our study confirms that a recessive distal titinopathy with young or early‐adult onset occurs from either two truncating mutations in the final two exons (363–364) or a combination of one mutation in these exons and a truncating mutation in a canonical exon on the other allele. In line with previous studies, our cohort also confirms the predominant lower limb involvement, the presence of a normal to mildly elevated serum creatine kinase level, and, above all, the lack of cardiac and respiratory involvement. Interestingly, all the patients we report here show marked asymmetric muscle involvement, that has been similarly observed in 5/14 patients with the Serbian founder mutation.
However, our study also confirms that truncating variants in exon 363 may also result in early‐onset myopathies in the presence of a second variant in exons with specific exon usage.
Author Contributions
Veronica Sian: conceptualization, data curation, writing‐original draft, writing – review and editing. Maria Francesca Di Feo: conceptualization, data curation, writing – review and editing. Sergei Kurbatov: investigation, resources, writing – review and editing. Anna Vihola: investigation, writing – review and editing. Helena Luque: investigation, writing – review and editing. Fedor Konovalov: investigation, resources, writing – review and editing. Stojan Peric: investigation, resources, writing – review and editing. Cathrina Duffy: investigation, resources, writing – review and editing. Cornelia Kornblum: investigation, resources, writing – review and editing. Kristl G. Claeys: investigation, resources, writing – review and editing. Peter Hackman: conceptualization, writing – review and editing. Bjarne Udd: conceptualization, investigation, supervision, writing – review and editing. Marco Savarese: conceptualization, data curation, supervision, writing‐original draft, writing – review and editing.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Table S1: Exon usage, expressed as percentage spliced in (PSI), of each mutated exon in fetal and adult muscles.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1M. Krüger and S. Kötter , “Titin, a Central Mediator for Hypertrophic Signaling, Exercise‐Induced Mechanosignaling and Skeletal Muscle Remodeling,” Frontiers in Physiology 7 (2016): 76, 10.3389/fphys.2016.00076.26973541 PMC 4771757 · doi ↗ · pubmed ↗
- 2S. Labeit and B. Kolmerer , “Titins: Giant Proteins in Charge of Muscle Ultrastructure and Elasticity,” Science 270, no. 5234 (1995): 293–296, 10.1126/science.270.5234.293.7569978 · doi ↗ · pubmed ↗
- 3D. O. Fürst , M. Osborn , R. Nave , and K. Weber , “The Organization of Titin Filaments in the Half‐Sarcomere Revealed by Monoclonal Antibodies in Immunoelectron Microscopy: A Map of Ten Nonrepetitive Epitopes Starting at the Z Line Extends Close to the M Line,” Journal of Cell Biology 106, no. 5 (1988): 1563–1572, 10.1083/jcb.106.5.1563.2453516 PMC 2115059 · doi ↗ · pubmed ↗
- 4M. Savarese , J. Sarparanta , A. Vihola , B. Udd , and P. Hackman , “Increasing Role of Titin Mutations in Neuromuscular Disorders,” Journal of Neuromuscular Diseases 3, no. 3 (2016): 293–308, 10.3233/JND-160158.27854229 PMC 5123623 · doi ↗ · pubmed ↗
- 5M. L. Bang , T. Centner , F. Fornoff , et al., “The Complete Gene Sequence of Titin, Expression of an Unusual ≈700‐k Da Titin Isoform, and Its Interaction With Obscurin Identify a Novel Z‐Line to I‐Band Linking System,” Circulation Research 89, no. 11 (2001): 1065–1072, 10.1161/hh 2301.100981.11717165 · doi ↗ · pubmed ↗
- 6A. Biquand , S. Spinozzi , P. Tonino , et al., “Titin M‐Line Insertion Sequence 7 Is Required for Proper Cardiac Function in Mice,” Journal of Cell Science 134, no. 18 (2021): jcs 258684, 10.1242/jcs.258684.34401916 PMC 8466004 · doi ↗ · pubmed ↗
- 7B. Kolmerer , N. Olivieri , C. C. Witt , B. G. Herrmann , and S. Labeit , “Genomic Organization of M Line Titin and Its Tissue‐Specific Expression in Two Distinct Isoforms,” Journal of Molecular Biology 256, no. 3 (1996): 556–563, 10.1006/jmbi.1996.0108.8604138 · doi ↗ · pubmed ↗
- 8K. Charton , L. Suel , S. F. Henriques , et al., “Exploiting the CRISPR/Cas 9 System to Study Alternative Splicing In Vivo: Application to Titin,” Human Molecular Genetics 25, no. 20 (2016): 4518–4532, 10.1093/hmg/ddw 280.28173117 · doi ↗ · pubmed ↗